Establishment of RIG-I knockout 293T cell line and its effect on the replication of influenza B virus
1. Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China;
2. University of Chinese Academy of Sciences, Beijing 100049, China;
3. College of Animal Science and Technology, Guangxi University, Nanning 530004, Guangxi, China;
4. China Institute of Veterinary Drug Control, Beijing 100081, China;
5. The High School Affiliated to Beijing Normal University, Beijing 100052, China
Received: May 7, 2019; Accepted: September 3, 2019; Published: September 18, 2019
Supported by: National Natural Science Foundation of China (No. 31672531), National Science and Technology Major Project (No. 2018ZX10101004)
CRISPR (Clustered regularly interspaced short palindromic repeats)/Cas系统是广泛存在于细菌与古菌中的具有抗感染作用的串联重复DNA序列,对防御外来噬菌体或质粒的入侵发挥着重要作用[1]。CRISPR最早在1987年由日本科学家于大肠杆菌的碱性磷酸酶基因侧翼中首次发现[2],直到2002年科学家们才将其正式命名为CRISPR[3]。已鉴定的CRISPR-Cas系统有3种,其中以Ⅱ型最简单且研究得最为透彻深入[4-5]。CRISPR中的重复序列一般为21–48 bp,间隔序列一般为21–72 bp (一般来源于外界入侵的DNA),CRISPR基因座在转录后进行加工生成成熟的crRNA。而编码具有核酸酶和解旋酶活性的Cas蛋白的基因则位于临近CRISPR的区域,Cas蛋白在crRNA的介导下识别靶向的DNA序列,对其进行切割,发挥免疫防御的功能[6-7]。基于CRISPR/Cas系统的作用原理,研究人员开发了通过人工合成sgRNA序列介导Cas蛋白对目的基因进行识别并剪辑的精准基因编辑技术。由于该技术具有高效性、廉价性且易于操作,目前已经被用于多种生物及细胞的基因编辑[8, 12]。
模式识别受体中的RIG-I样受体是定位于细胞质中的一类RNA解旋酶,它们可以识别入侵的含5′-三磷酸基团病毒RNA,触发下游信号分子的活化,进而导致干扰素及炎性因子的产生,以抵抗病毒的入侵。RIG-I的结构包括N端的2个CARD结构域(Caspase recruiting domain, CARD)、中间的1个DExD/H解旋酶结构域和C端的1个抑制区域(Repressor domain,RD)。RIG-I的3个结构域对维持RIG-I的功能都具有重要作用,CARD结构域负责下游信号的转导,DExD/H结构域具有ATPase活性和RNA解旋酶功能,而RD结构域则负责与病毒RNA的5′-三磷酸基团结合[13-14]。在没有5′-三磷酸基团存在的情况下,RIG-I以“静息”的状态存在,其CARD结构域和RD结构域“绑定”在一起。而当病毒RNA的5′-三磷酸基团与RIG-I的C端的RD结构域结合后,RIG-I构象则发生改变,CARD与RD分开,RIG-I变为开放状态,开放的RIG-I与其接头蛋白MAVS结合,诱导下游的IFN等因子表达[15-16]。RIG-I可识别多种病毒的RNA,其中仙台病毒(Sandai virus,SeV)是经典的通过激活RIG-I信号通路介导大量干扰素产生的模式病毒[17, 24]。
研究表明RIG-I识别A型流感病毒的ssRNA后,引发RIG-I与MAVS结合,引起干扰素调节因子IFR3/7的活化和NF-κB的入核,最终诱导干扰素及炎性因子等产生,从而抑制病毒的复制与传播[18, 25-26]。前期研究发现,B型流感病毒(IBV)在感染早期能上调细胞中RIG-I的蛋白表达水平,为了探索RIG-I是否为B型流感病毒激活下游信号通路的主要受体及其对IBV复制的影响,我们构建并使用RIG-I敲除的293T细胞系证明了RIG-I信号通路是宿主抵抗IBV感染的主要天然免疫通路之一,RIG-I基因敲除后对IBV在293T细胞中的复制具有促进作用,为探索IBV的感染机制奠定了基础。
1 材料与方法
1.1 材料
1.1.1 细胞株和质粒 293T细胞系和MDCK细胞系均由本实验室保存,UCATM CRISPR/Cas9快速构建及活性检测试剂盒购自北京百奥赛图生物技术有限公司,大肠杆菌DH5α感受态细胞由本实验室自制和保存,B型流感病毒(B/Shanghai/PD114/2018,Yamagata lineage,IBV)于2018年分离自上海浦东的一位B型流感病毒患者,由国家流感中心的王大燕教授馈赠。仙台病毒(Sandai Virus,SeV)由本实验室保存。
1.1.2 实验试剂 NEB buffer2与限制性内切酶BbsⅠ购自New England Biolabs公司。转染试剂Lipofectamine 2000 Reagent及琼脂糖购自Invitrogen公司。寡核苷酸回收试剂盒购自北京艾莱德生物技术有限公司。胎牛血清及DMEM培养基购自Gibco公司。高保真DNA聚合酶购自北京博迈德生物技术有限公司。质粒提取试剂盒、血液/细胞/组织基因组DNA提取试剂盒购自北京天根生物技术有限公司。Anti-RIG-I (D14G6) mAb、anti-IRF3 mAb和anti-P-IRF3 Ab购自Cell Signaling Technology公司,anti-p65 mAb和anti-P-p65 mAb购自Santa Cruz公司,Trizol Reagent购自Life Technology公司,SYBR® Premix Ex TaqTM kit购自TaKaRa公司。
1.1.3 引物合成及PCR检测产物的测序 所有引物合成及PCR产物测序均由北京博迈德生物技术有限公司完成。
1.2 实验方法
1.2.1 sgRNA的设计与寡核苷酸链的合成 从GenBank数据库中下载人类的RIG-I (Gene ID登录号:23586)全基因序列,并标识出其基因编码区(Coding sequence, CDS)。利用DNA Star软件对CDS区域进行GG(N)18NGG基序的查找,以查找到的符合要求的基序作为sgRNA的正链,且在其正链的5′端添加CACC,在其负链的5′端添加AAAC,以便与BbsⅠ酶切后的载体上的粘性末端互补,sgRNA的核苷酸序列如表 1所示。
表 1 sgRNA的核苷酸序列
Table 1 Nucleotide sequences of sgRNAs
| Name |
Sequence (5′–3′) |
| Hu-RIG-I-sgRNA1-F |
ACCGGATTATATCCGGAAGACCC |
| Hu-RIG-I-sgRNA1-R |
AAACGGGTCTTCCGGATATAATCC |
| Hu-RIG-I-sgRNA2-F |
CACCGGCAGGTGCAGAGAAATTGG |
| Hu-RIG-I-sgRNA2-R |
AAACCCAATTTCTCTGCACCTGCC |
| Hu-RIG-I-sgRNA3-F |
CACCGGAACAAGTTCAGTGAACTG |
| Hu-RIG-I-sgRNA3-R |
AAACCAGTTCACTGAACTTGTTCC |
1.2.2 重组Hu-RIG-I-sgRNA-pCS-1载体构建 将合成的单链sgRNA序列退火,使其形成粘末端双链。退火体系为:Hu-RIG-I-sgRNA-F 20 μL,Hu-RIG-I-sgRNA-R 20 μL,NEB buffer 2 10 μL;退火条件为:于PCR仪中97 ℃作用7 min,然后关机,自动降温1 h后取出样品进行利用寡核苷酸回收试剂盒进行胶回收纯化。将回收后具粘性末端的双链片段与BbsⅠ内切酶线性化后的pCS-1质粒1–2 μg在16 ℃下连接4 h,连接体系为:双链片段6 μL,pCS-1质粒2 μL,T4 DNA连接酶1 μL,T4 DNA连接酶缓冲液1 μL。将连接产物转化至感受态E. coli DH5α中,氨苄抗性筛选阳性克隆并挑取阳性单克隆接种氨苄抗性的液体LB培养基,利用质粒提取试剂盒进行质粒提取并测序验证是否为正确的重组质粒。
1.2.3 重组pUCA(Luc)-target载体的构建 以293T细胞基因组DNA为模板,利用高保真DNA聚合酶扩增sgRNA靶位点上下游间的一段长约400–500 bp的序列作为target,扩增引物如表 2所示。PCR产物测序验证后与线性化的precut pUCA (Luc)质粒连接,构建重组载体pUCA (Luc)-target,以用于Hu-RIG-I-sgRNA-pCS-1的向导及切割(sgRNA/Cas9)活性检测。连接体系如下:纯化的PCR片段(target) 4 μL,precut pUCA(Luc)质粒1 μL,T4 DNA连接酶1 μL,T4 DNA连接酶缓冲液1 μL,ddH2O 3 μL。连接条件为16 ℃连接过夜。将连接产物转化至感受态细胞E. coli DH5α中,卡那抗性筛选阳性克隆并接种至卡那抗性的液体LB培养基,利用质粒提取试剂盒进行质粒提取并测序验证是否为正确的重组质粒。
表 2 扩增Target序列的引物
Table 2 Primers for target segments amplification
| Primer name |
Primer sequence (5′–3′) |
| Targeting-sgRNA1-F |
TAAAGCTAGTGAGGCACAGCCT |
| Targeting-sgRNA1-R |
CACCTCGCTGGAACTCAGTTT |
| Targeting-sgRNA2+ sgRNA3-F |
CATGGATATGACCCACTGAGCT |
| Targeting-sgRNA2+ sgRNA3-R |
CTTTGGGCCAGATGGGCTAAT |
1.2.4 Hu-RIG-I-sgRNA-pCS-1的活性检测 将生长状态良好的293T细胞分至96孔板,待细胞汇合度达到80%时换液至OPTI-MEM培养基中,1–2 h后转染Hu-RIG-I-sgRNA-pCS-1和Seq-sgRNA-pUCA (Luc)质粒,转染量分别为80 ng/孔和20 ng/孔,所用转染试剂为Lipofectamine 2000 Reagent,质粒与转染试剂的比例为1:2.5,同时设立空白对照、阴性对照和阳性对照,空白对照不加任何质粒,阴性对照组只加pUCA (Luc)-target质粒,阳性对照加pUCA (Luc)-target和pCS-positive质粒,每组设立3个重复孔。转染后4–6 h换液至含10% FBS的DMEM培养基进行培养,24 h后进行萤光素酶活性检测,具体步骤按试剂盒说明书进行,Luc反应液及参数设置按说明书进行配制及设定。
1.2.5 293T细胞的嘌呤霉素药物致死浓度筛选 将生长状态良好的293T细胞分至24孔板,待细胞长至100%汇合度时加入不同浓度的嘌呤霉素进行药物致死浓度的筛选。所用药物浓度梯度为:0、0.5、1.0、1.5、2.0、2.5、3.0、3.5、4.0、4.5、5.0、6.0 μg/mL,每个浓度设立2个孔。每天观察细胞死亡情况,挑取导致细胞在5 d内全部死亡的最低剂量的药物浓度作为本实验的药物筛选浓度。
1.2.6 高活性Hu-RIG-I-sgRNA-pCS-1质粒的细胞转染 将生长状态良好的293T细胞分至6孔板,待细胞长至80%汇合度时换液至OPTI-MEM中,1–2 h后转染活性较高的Hu-RIG-I-sgRNA-pCS-1质粒,每孔的转染量为2 μg,所用转染试剂为Lipofectamine 2000 Reagent,其与质粒的比例为1:2.5,同时转染pCS-1空载体作为阴性对照。转染后4–6 h换液至含10% FBS的DMEM培养基进行培养。
1.2.7 药物筛选及单克隆培养 将转染后换液至10% FBS的DMEM培养基中的293T继续培养12 h后开始在培养基中加入嘌呤霉素进行筛选,每24 h更换1次培养基。筛选3 d后嘌呤霉素的用量减半,继续筛选3 d后更换为无嘌呤霉素的含10% FBS的DMEM培养基,根据细胞生长情况继续培养2–3 d后利用流式细胞仪进行单细胞分选至96孔板,每孔150 μL培养基。7–10 d后,将细胞单克隆消化分至24孔板,并逐渐扩大培养,以便下一步PCR鉴定阳性克隆。
1.2.8 PCR鉴定阳性克隆 将得到的单克隆用基因组提取试剂盒进行基因组DNA的提取作为模板,PCR扩增sgRNA靶位点上下游区间的DNA序列,检测敲除效果。PCR扩增所用引物序列见表 2。PCR反应体系为:模板DNA 5 μL,Taq Mix 12.5 μL,Primer-F 1 μL,Primer-R 1 μL,ddH2O 5.5 μL。PCR反应程序为:94 ℃预变性5 min;94 ℃变性30 s,58 ℃退火30 s,72 ℃延伸1 min,40个循环;然后72 ℃延伸10 min;4 ℃下保存。PCR结束后取10 μL的反应产物进行琼脂糖凝胶电泳检测。
1.2.9 Western blotting鉴定阳性克隆 将PCR鉴定阳性的单克隆及野生型(wild type, WT)的293T细胞分至6孔板,待细胞铺满后,换液至无血清的DMEM培养基中,用IBV及SeV进行感染,MOI=0.5,于0、4、8、12、24 h定点取样,以细胞裂解液进行裂解,裂解产物采用BCA法蛋白定量后进行Western blotting,检测RIG-I蛋白的表达情况。
1.2.10 RIG-I对IBV诱导的干扰素、炎性因子及干扰素刺激基因的影响 将生长状态良好的WT及RIG-I-/- 293T细胞铺至12孔板,待细胞密度达到100%时感染IBV,MOI为0.5。分别于感染后0、4、8、12 h取样,每孔加入Trizol Reagent 500 μL裂解细胞并进行RNA提取及反转录。反转录体系为:RNA模板5 μL,10 mmol/L dNTPs 1 μL,Oligo(dT) 1 μL,RRI 1 μL,MLV反转录酶1 μL,10×缓冲液2 μL,ddH2O 9 μL;反应条件为:RNA模板与Oligo(dT)混匀在70 ℃下作用5 min,冰浴5 min,再加入其他成分,42 ℃作用60 min,80 ℃条件下10 min即可。将所得DNA模板置于−20 ℃冰箱中冻存备用。Real-time qPCR的详细操作步骤按SYBR® Premix Ex TaqTM kit说明书进行,所用引物如表 3所示。
表 3 细胞因子的Real-time PCR引物
Table 3 Real-time PCR primers for detecting cytokines
| Primer name |
Primer sequence (5′–3′) |
| IFN-α-F |
CTGAATGACTTGGAAGCCTG |
| IFN-α-R |
ATTTCTGCTCTGACAACCTC |
| IFN-β-F |
TAGCACTGGCTGGAATGAGA |
| IFN-β-R |
TCCTTGGCCTTCAGGTAA TG |
| IFN-λ2-F |
CTCAGGTTGCATGACTGGTGG |
| IFN-λ2-R |
GAGGCC TCTGTCACCTTCAAC |
| IFN-λ3-F |
CAGCTGCAGGTGAGGGAGCGCCCCG |
| IFN-λ3-R |
GGTGGCCTCCAGAACCTT |
| IFN-λ1-F |
GGACGCCTTGG AAGAGTCACT |
| IFN-λ1-R |
AGAAGCCTCAGGTCCCAATTC |
| IL-6-F |
AGCCACTCACCTCTTCAGAACGAA |
| IL-6-R |
CAGTGCCTCTTTGCTGCTTTCACA |
| TNF-α-F |
TGGGCTCCCTCTCATCAGTTC |
| TNF-α-R |
TCCGCTTGGTGGTTTGCTAC |
| CXCL10-F |
CCAAGTGCTGCCGTCATTTTC |
| CXCL10-R |
GGCTCGCAGGGATGATTTCAA |
| MxA-F |
GGTGGCTGAGAACAACCTGT |
| MxA-R |
GGTCCTGCTCCACACCTAG A |
| IFIT2-F |
AAGCACCTCAAAGGGCAAAAC |
| IFIT2-R |
TCGGCCCATGTGATAGTAGAC |
| IFIT3-F |
TCAGAAGTCTAGTCACTTGGGG |
| IFIT3-R |
ACACCTTCGCCCTTTCATTTC |
| ISG20-F |
CGACACGTCCACTGACAGGCTGTTG |
| ISG20-R |
TCCATCGTTGCCCTCGCATCTTC |
1.2.11 RIG-I对IBV感染后p65、磷酸化P65、IRF3及磷酸化IRF3表达的影响 将生长状态良好的WT及RIG-I-/- 293T细胞铺至6孔板,待细胞密度达到100%时感染IBV,同时以SeV感染作为对照,MOI均为0.5。分别于感染后0、4、8、12、24 h时用细胞裂解液裂解细胞,利用相应抗体对裂解液中的p65、P-p65、IRF3及P-IRF3蛋白进行Western blotting检测。
1.2.12 RIG-I对IBV生长曲线的影响 将生长状态良好的WT及RIG-I-/- 293T细胞铺至12孔板,待细胞密度达到100%时感染IBV,MOI为0.5。分别于感染后0、12、24、36、48、60、72 h收取细胞上清,进行病毒TCID50的测定。简要步骤如下:将生长状态良好的MDCK细胞铺至96孔板;用无血清DMEM 4倍梯度稀释不同时间点的细胞上清;待MDCK细胞密度达到100%时,弃去培养基,用PBS洗涤,然后将稀释后的细胞上清加入孔中,每孔200 μL,每个稀释度10个重复孔,同时设立阴性对照孔(只加无血清DMEM);培养72 h后观察细胞病变情况,计算不同时间点收取的细胞上清的TCID50,绘制并比较IBV在两种细胞系上的生长曲线。
2 结果与分析
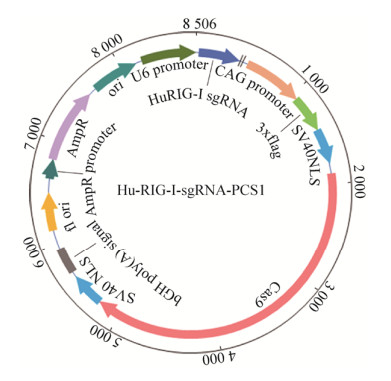
2.1 重组载体Hu-RIG-I-sgRNA-pCS-1的构建 RIG-I的CARD结构域负责下游信号的转导,对RIG-I功能的发挥具有至关重要的作用,若对CARD结构域基因进行缺失,RIG-I信号转导通路则被阻断。而CARD结构域的基因位于Exon1– Exon4上,于是我们针对Exon1和Exon4设计3个不同的sgRNA寡核苷酸单链(Hu-RIG-I-sgRNA1–3),其中Hu-RIG-I-sgRNA1位于Exon1上,Hu-RIG-I-sgRNA2–3位于Exon4上。合成的寡核苷酸单链被引入了BbsⅠ酶切位点,因此可通过退火形成具有粘性末端的双链,然后与经过BbsⅠ酶切的pCS-1载体连接,构成重组质粒Hu-RIG-I-sgRNA-pCS-1,构建示意图如图 1所示。重组质粒Hu-RIG-I-sgRNA-pCS-1可转录靶向目的基因的sgRNA,同时该质粒还可表达Cas9蛋白,因此该质粒可同时完成靶向与切割的功能。pCS-1载体骨架中的嘌呤霉素抗性基因可用于阳性克隆的筛选。
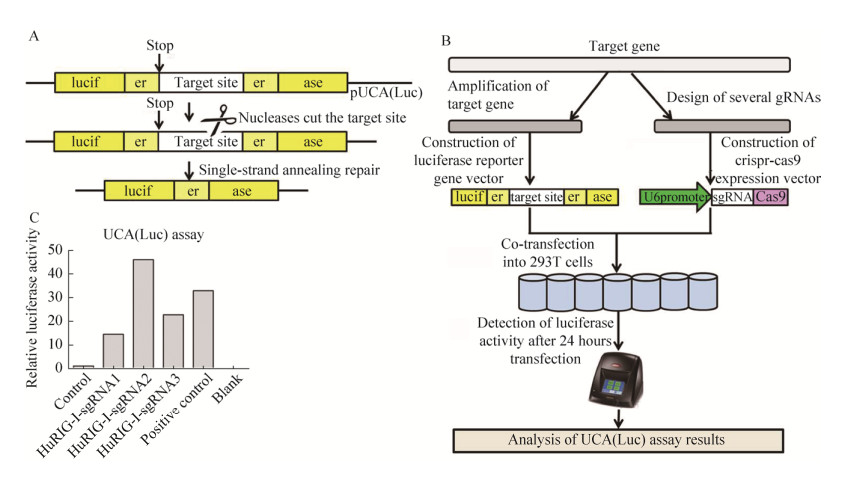
2.2 Hu-RIG-I-sgRNA-pCS-1向导及切割活性的检测 如图 2A所示,pUCA(Luc)是sgRNA的报告基因质粒,其中的荧光素酶(Luciferase,Luc)基因内部含有终止密码子和CRISPR/Cas9的靶位点序列。终止密码子和靶位点序列两端的“er”为荧光素酶的互补重复序列。终止密码子可导致荧光素酶的翻译提前终止,表达无功能的荧光素酶。但若CRISPR/Cas9对靶位点进行了切割,则可引发基于SSA (Single strand annealing)的DNA同源重组修复机制,互补重复序列“er”通过同源重组而形成了完整的荧光素酶编码序列,从而表达有功能的荧光素酶,荧光素酶的活性与sgRNA/ Cas9的活性呈正相关。同时转染pUCA(Luc)-target与Hu-RIG-I-sgRNA-pCS-1质粒,通过荧光素酶活性判断Hu-RIG-I-sgRNA-pCS-1质粒的sgRNA/ Cas活性。在进行活性检测时,会使用阳性对照质粒(pCS-Positive质粒, 该质粒可表达一段已验证过具有活性的positive-sgRNA,序列为:5′-GGC TAATAACTTAATCGTGG-scaffold-3′)作为参考。precut pUCA (Luc)质粒的“er”序列间具有positive-sgRNA的靶位点序列,且克隆为环状质粒后仍然存在。因此在转染实验中,转染pUCA (Luc)-target和pCS-Positive质粒即可作为活性检测实验的阳性对照,活性检测流程如图 2B所示。荧光素酶活性检测结果如图 2C所示,从图 2C可以看出,Hu-RIG-I-sgRNA2-pCS-1的活性最高。
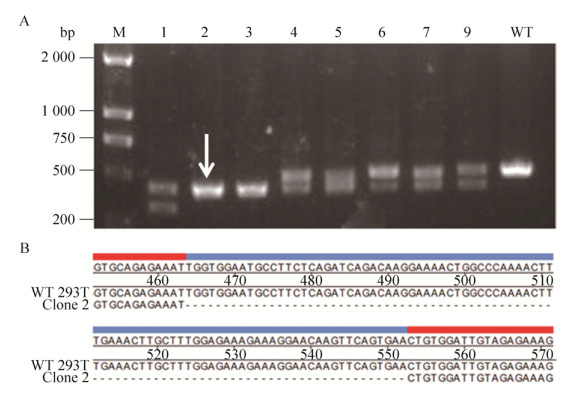
2.3 重组载体Hu-RIG-I-sgRNA2-pCS-1可高效切割靶位点造成RIG-I基因部分缺失 选取导致293T细胞在5 d全部死亡的嘌呤霉素浓度3.0 μg/mL作为本实验的药物筛选浓度。然后根据2.2实验结果,挑取sgRNA活性最高的重组质粒Hu-RIG-I-sgRNA2-pCS-1转染293T细胞,并于加入嘌呤霉素后开始每天观察细胞的死亡情况。结果表明,细胞于加入嘌呤霉素后3 d开始出现大量的死亡,5 d后嘌呤霉素的浓度降为1.5 μg/mL,再筛选3 d后撤除嘌呤霉素压力,继续培养5–7 d后可明显看到细胞形成白色单克隆,将细胞进行消化后制成悬液,用流式细胞仪进行单细胞分选,获得单克隆。对获得的单克隆进行PCR鉴定,结果表明,重组质粒Hu-RIG-I-sgRNA2-pCS-1转染至293T细胞后,可对细胞基因组靶位点进行高效切割,造成全部或部分等位基因的基因敲除。对筛选到的22株单克隆进行基因组DNA提取和PCR检测,共筛选到8株疑似阳性克隆,如图 3所示。从图中可以看出,1号克隆出现两条较对照小的目的条带,可能是因为不同的等位基因发生删减的长度不同。而4、5、6、7、8号克隆也出现了大小不同的两条带,经测序发现,分子量较大的条带序列与野生型293T细胞的序列相同,没有发生删减。2、3号克隆均为单一条带,与野生型293T细胞的序列相比,明显变小,说明所有等位基因均发生了相同的敲除,经测序发现2、3号克隆缺失序列均为CDS区域的第464–552位核苷酸,共89个核苷酸,而氨基酸的密码子则从155位开始移码并发生缺失突变,且突变后的核苷酸序列中提前出现了终止密码子(第166位氨基酸)。由此推断2、3号克隆无法表达具有正常生物学功能的RIG-I蛋白,挑取2号克隆用于后续实验,命名为RIG-I-/- 293T细胞,并进行功能实验验证。
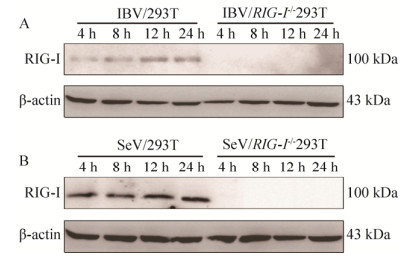
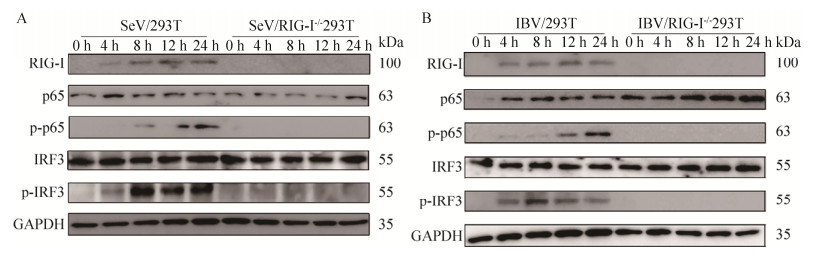
2.4 IBV及SeV感染RIG-I-/- 293T细胞后RIG-I蛋白表达缺失 通常RIG-I在绝大多数组织的细胞中有少量表达,在病毒的刺激下其表达会迅速上调[26]。用细胞裂解液对IBV感染后不同时间的细胞进行裂解,以Anti-RIG-I的单抗(以RIG-I蛋白C端的652–925位氨基酸部分作为抗原进行免疫获得)进行RIG-I蛋白的Western blotting检测。SeV作为经典研究RIG-I信号通路的模式病毒,其感染后可激活细胞中RIG-I介导的RLR相关天然免疫通路,引起RIG-I蛋白表达上调及各种细胞因子的表达[27],因此选用SeV感染作为参考,检测RIG-I-/- 293T细胞中RIG-I的内源性表达情况。结果如图 4所示,无论是IBV还是SeV,病毒感染后RIG-I-/- 293T细胞中均检测不到RIG-I蛋白的表达,而野生型293T细胞在二者感染后的4–24 h均可检测到不同程度的RIG-I蛋白的表达,表明RIG-I-/- 293T细胞中RIG-I基因敲除成功。
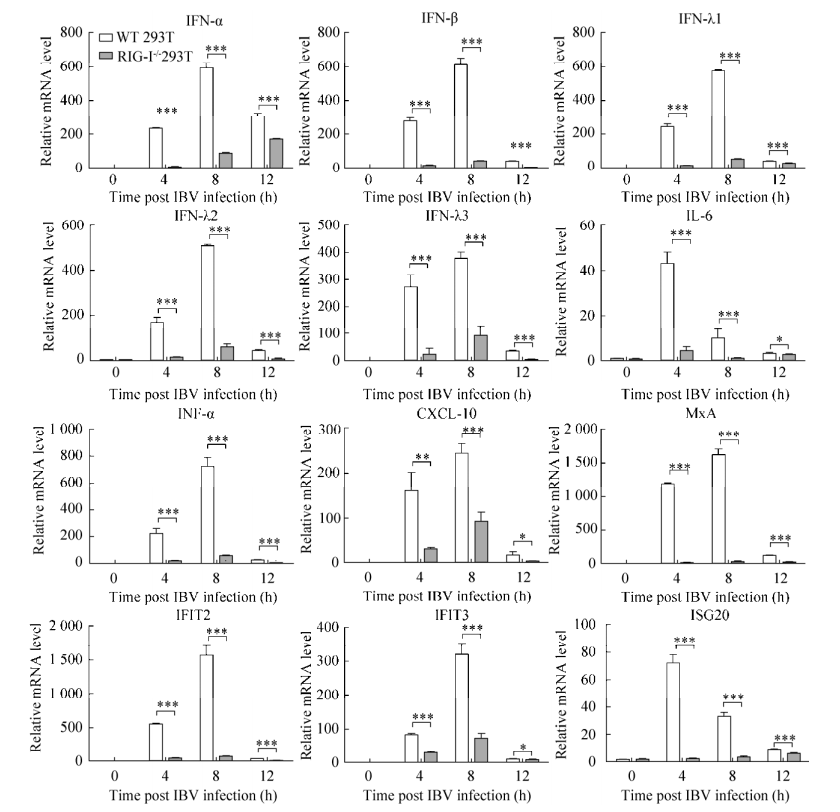
2.5 RIG-I可上调IBV诱导的干扰素、炎性因子及其干扰素刺激基因的转录 当带有5′-三磷酸基团的病毒RNA作为配体与RIG-I结合后,RIG-I被激活,进而与其下游的接头蛋白MAVS结合,接着通过一系列的信号转导诱导Ⅰ型干扰素的产生[28]。干扰素与细胞表面的干扰素受体结合后则启动JAK-STAT途径,起始干扰素诱导基因的转录表达,发挥抗病毒作用[29]。为了验证RIG-I是否为IBV激活天然免疫通路的主要受体和RIG-I对IBV感染后干扰素、炎性因子及干扰素刺激基因表达的影响,我们将IBV对WT及RIG-I-/- 293T同时进行了感染,并检测干扰素、炎性因子及干扰素刺激基因的转录水平在两种细胞中的差异。如图 5所示,在IBV感染后的RIG-I-/- 293T细胞中,IFN-β、IFN-λ1、IFN-λ2、IL-6、TNF-α、MxA、IFIT2和ISG20的转录水平明显低于野生型293T细胞,而IFN-α、IFN-λ3、CXCL-10和IFIT3的转录水平与未感染时比,虽然有一定程度的上调(可能是RIG-I信号通路被阻断后,其他信号通路进行了部分代偿),但仍明显低于野生型293T细胞,说明RIG-I是介导IBV诱导的干扰素、炎性因子及干扰素刺激基因大幅上调的主要受体。
RNA病毒感染细胞后,可引起较高水平的Ⅰ型干扰素表达[30],而在本实验中发现,IBV诱导后,野生型293T细胞中还检测到较高水平的Ⅲ型干扰素的mRNA的转录水平(即IFN-λ1、IFN-λ2和IFN-λ3),暗示了Ⅲ型干扰素在机体抵抗病毒IBV入侵的过程中也发挥着相当重要的作用。
2.6 RIG-I是IBV激活抗病毒天然免疫信号通路的主要受体之一 RIG-I介导信号转导的一个关键的步骤是IRF3/7和NF-κB的磷酸化[28]。p65是NF-κB蛋白家族的一个重要成员,p65与p50的二聚体形式是最常见的NF-κB二聚形式,因此p65的磷酸化是NF-κB活性形式的基础。在本研究中,将IBV对WT及RIG-I-/- 293T进行感染并检测p65、P-p65、IRF3及P-IRF3的表达(图 6B),同时采用SeV感染作为参考(图 6A)。从图中可以看出,IBV及SeV诱导的p65及IRF3在WT及RIG-I-/- 293T细胞系中的表达没有明显差异,而P-p65及P-IRF3在野生型293T细胞中可以检出,在RIG-I-/- 293T细胞中完全没有表达,说明RIG-I敲除后并不影响p65及IRF3的表达,但可抑制二者的磷酸化。不管病毒感染与否,RIG-I-/- 293T细胞中都检测不到P-p65及P-IRF3的发生,表明IBV与SeV相似,其感染诱导的干扰素、炎性因子及其干扰素刺激基因的产生与RIG-I介导的抗病毒天然免疫反应密切相关。
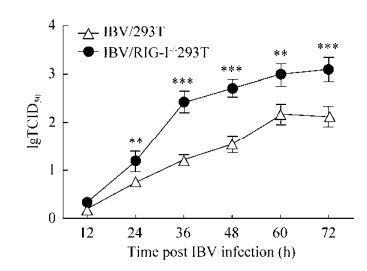
2.7 RIG-I抑制IBV在293T细胞中的复制 通过2.5的结果可知,RIG-I可上调IBV诱导的干扰素、炎性因子和干扰素刺激基因等细胞因子在293T细胞中的转录水平,而这些细胞因子对宿主抵抗病毒的入侵具有非常重要的作用,这说明RIG-I在宿主细胞抵抗IBV感染的过程中发挥着重要作用。为了进一步探索RIG-I对IBV的生长复制的影响,我们绘制了IBV在野生型及RIG-I-/-293T细胞中的多步生长曲线(图 7)。从图中可以看出,IBV在RIG-I-/- 293T细胞中的lgTCID50显著高于其在野生型293T细胞中的lgTCID50,这说明RIG-I对IBV在293T细胞中的复制具有抑制作用,敲除RIG-I基因可提高IBV在293T细胞中的复制水平。
3 讨论 高效精准的基因编辑技术是研究基因功能的重要工具,CRISPR/Cas9基因编辑技术是基于细菌CRISPR/Cas9免疫系统的可在基因组特定位点进行靶向编辑(缺失或插入)的新技术[11-12]。CRISPR/Cas9免疫系统的作用机制分为CRISPR间隔序列的获得、crRNA前体的转录与加工、crRNA、tracrRNA及Cas9蛋白形成复合体对外源性DNA进行剪切加工[5, 31]。以该机制为基础,研究人员开发出CRISPR/Cas9介导的基因编辑技术,根据成熟crRNA和其互补tracrRNA的二聚体结构设计出sgRNA,使之与Cas9蛋白结合,引导Cas9蛋白与目的基因位点结合并进行剪辑[31]。该设计将CRISPR/Cas9免疫系统简化为sgRNA与Cas蛋白的复合体,将该复合体导入细胞即可对完成对目的基因的切割。针对不同的靶点只需替换20多个碱基长短的sgRNA即可,使得基因编辑易于操作,大大地简化了工作流程。该技术简单快捷、高效精准,已应用于多种细胞及哺乳动物(大鼠、小鼠、恒河猴等)的基因编辑,为人类疾病(如癌症等)及基因未知功能的研究提供了大大的便利[32, 35]。
IBV最早于1940年分离到,属于具囊膜的单股负链RNA病毒,其基因组由8个节段组成,可引起局部流感的发生[36-37]。IBV的自然宿主为人,在海洋哺乳动物海豹中也曾检测到[38]。IBV基因组的变异率及重组率均低于A型流感病毒(IAV),因此学界一直认为IBV的危害要远远小于IAV,因此关于IBV的研究相对较少。但近年来,IBV在某些特定的环境下也造成了不亚于IAV的危害,例如2013–2014年IBV在北美特别是美国造成了大规模的疫情,2017年冬至2018年春,IBV的Yamagata谱系与Victoria谱系在中国的南北方交替流行,造成了数万人感染及沉重的疾病负担[39]。因此IBV的致病性是不可低估的,研究IBV的致病机理也是刻不容缓的,并且本实验中所用毒株为2017年B型流感暴发时收集的临床毒株,从而使本实验更具有临床研究意义,并对今后的B型流感的相关研究及预测打下基础。
为了探索RIG-I是否为IBV激活下游信号通路的主要受体及其对IBV复制的影响,我们进行了RIG-I敲除的293T细胞系的构建。在分析了RIG-I的不同结构域的不同功能之后,设计了针对RIG-I蛋白N端的CARD结构域的sgRNA,这样可以使切割发生在RIG-I的N端,加上可能发生移码突变,使RIG-I蛋白表达受阻,达到敲除的目的。本研究针对RIG-I基因的敲除发生在CARD结构域,共有89个核苷酸的删减,且造成了氨基酸从155位开始移码,且终止于第166位氨基酸,使得细胞无法表达RIG-I蛋白,为研究RIG-I是否为B型流感病毒激活下游信号通路的主要受体及其对IBV复制的影响提供了基础。
目前关于B型流感病毒与天然免疫通路的关系的研究主要集中在NS1蛋白对宿主细胞IFN产生的影响上,而关于哺乳动物细胞天然免疫系统是如何对抗B型流感病毒的研究尚少。我们通过实验发现IBV感染哺乳动物细胞后,RIG-I的表达迅速上调,且可诱导IFN等细胞因子的快速产生,而敲除细胞中的RIG-I基因后,IFN等细胞因子的转录水平显著下降,该结果说明IBV诱导的IFN等细胞因子的产生具有一定程度的RIG-I信号通路依赖性。进一步研究发现,IBV感染后,RIG-I敲除导致其信号通路下游的转录因子IRF3及NF-κB的亚基p65无法发生磷酸化,同时IBV的复制效率提高,说明RIG-I介导的抗病毒天然免疫信号通路的激活对IBV的复制具有抑制作用。以上研究为揭示IBV天然免疫学特征奠定了基础。
 2020, Vol. 36
2020, Vol. 36