 2021, Vol. 37
2021, Vol. 37中国科学院微生物研究所、中国微生物学会主办
文章信息
- 王梦男, 韩明飞, 刘炳辉, 田春艳, 朱云平
- Wang Mengnan, Han Mingfei, Liu Binghui, Tian Chunyan, Zhu Yunping
- 基于集成的共表达网络分析方法研究3种癌症的肿瘤相关模块
- Integration-based co-expression network analysis to investigate tumor-associated modules across three cancer types
- 生物工程学报, 2021, 37(11): 4111-4123
- Chinese Journal of Biotechnology, 2021, 37(11): 4111-4123
- 10.13345/j.cjb.210137
-
文章历史
- Received: February 10, 2021
- Accepted: March 11, 2021


引用本文

|



2. 军事兽医研究所, 吉林 长春 130000
2. Institute of Military Veterinary Medicine, Academy of Military Medical Sciences, Academy of Military Sciences, Changchun 130000, Jilin, China
基因往往不是独立发挥作用,而是多个基因相互协调,共同参与某一生物学过程。参与同一生物学过程的基因对或基因模块在不同样本中同时表现出上调或下调的趋势,称之为基因共表达现象。在此基础上,研究人员将疾病与健康状态相比新建立或失去共表达现象的基因模块,称为差异共表达(Differential co-expression,DC) 基因模块。在疾病中,新建立的共表达模块往往参与因疾病而激活的生物过程,而失去共表达趋势的模块往往参与在疾病中失调的生物过程。近年来,差异共表达分析已被广泛用于探索各类疾病的机制[1-10]。此外,差异表达(Differential expression,DE) 可以鉴定疾病与健康状态下表达水平显著变化的基因,因此一直是在分子水平上研究人类致病基因的重要手段。DC基因和DE基因均被证实与疾病的发生发展相关,但目前大部分研究仍集中在分别鉴定DC和DE基因[11-17],DC和DE分析未能得到有效的整合。尽管Lui等提出的DECODE方法可以整合单个基因的DC和DE特征来鉴定致病基因[18],但目前仍缺乏有效整合共表达模块的DC和DE基因特征的方法。
针对这一问题,我们开发了一种新的整合DC和DE特征的分析框架DC & DEmodule (图 1),并用于分析Gene Expression Omnibus (GEO)数据库中下载的6组肿瘤/癌旁微阵列(Microarray) 数据(肝癌、胃癌和结直肠癌各两组)。DC & DEmodule包含两个关键步骤:1) 分别鉴定肿瘤和癌旁组织的共表达基因模块;2) 整合DC和DE特征鉴定肿瘤相关模块,包括激活模块(肿瘤样本中上调且共表达增强) 和失能模块(肿瘤样本中下调且失去共表达)。

|
| 图 1 肿瘤相关模块鉴定的工作流程 Fig. 1 Schematic procedure for the identification of tumor-associated modules. We integrated two GEO microarray datasets associated with liver, gastric and colon cancer, respectively. For each cancer type, we used WGCNA to identify tumor/normal co-expression modules in each of the two datasets, then extracted the significantly conserved modules across individual datasets. We then identified the modules showing both differential expression levels and distinct co-expression strengths between normal and tumor conditions, including oncogenic modules (gaining co-expression and up-regulated in tumor) and dysfunctional modules (losing co-expression and down-regulated in tumor). |
| |
最终,我们在肝癌、胃癌和结肠癌中分别确定了2、5和2个激活模块以及5、5和1个失能模块,并且在3种癌症的激活模块中分别鉴定出17、69和11个关键基因。这些关键基因包含53个已报道的和3个新的癌症预后标志物TCEB1 (transcription elongation factor B (SIII),polypeptide 1 (15 kDa,elongin C)、RFC4 (Replication Factor C Subunit 4)和TRPC4AP(Transient Receptor Potential Cation Channel Subfamily C Member 4)。生存分析表明3个新标志物分别与肝癌、胃癌和结直肠癌的总体生存时间(Overall survival) 显著相关。随后,我们分别基于3种癌症的关键基因训练了3个随机森林分类器,用于区分TCGA和GEO数据库中的肿瘤和癌旁样本,结果显示分类的平均准确性达到93%。最后,我们利用独立的DC分析(基因集共表达分析,GSCA(Gene Set Co-expression analysis)、独立的DE分析(Student’s t-test)以及现有整合DC和DE分析的工具(DECODE) 分析3种癌症的表达谱,并与DC & DEmodule的结果比较。通路富集分析表明DC & DEmodule在检测已报道的肿瘤相关通路和发现新通路方面均具有更高的灵敏度。综上,我们的整合分析框架可以更好地发现肿瘤相关通路和生物标志物,并为DC和DE的联合分析提供新的视角。
1 材料与方法 1.1 材料从GEO数据库中(www.ncbi.nlm.nih.gov/geo)分别收集肝癌、胃癌和结直肠癌各两个微阵列表达谱。所有表达谱均含有从同一患者的肿瘤和相邻的癌旁组织切除的样本,包括GSE57957[19]、GSE17548[20]、GSE13911[21]、GSE19826[22]、GSE23878[23]和GSE41328[24]。根据以下流程对原始数据进行预处理:1) 进行RMA[25]数据标准化;2) 删除在所有样本中表达量空值比例大于20%的基因(当多个探针匹配到同一基因时,优先保留最大的表达值);3) 选取在同一癌症的两组微阵列数据中共同检测到的基因;4) 过滤掉在neXtprot[26]中不存在的基因。数据预处理结果见补充表S1。
1.2 方法 1.2.1 构建基因共表达网络应用R语言中WGCNA程序包对每个表达谱分别构建肿瘤和癌旁样本共表达网络(图 2步骤1),接着对网络进行层次聚类,然后对层次聚类树进行动态切割[27],将具有相似表达模式的基因归为一个共表达模块。为了整合每种癌症类型的两个独立的表达数据,我们取两组数据的所有模块组成两两模块对,使用费舍尔精确检验评估每个模块对中基因的保守性。当一个模块对的费舍尔精确检验P值小于0.05,且具有多于10个重叠基因时,这些重叠基因就组成两组数据的一个保守模块。最终我们鉴定到每种癌症高度保守的肿瘤/癌旁模块。模块结果见补充表S2。

|
| 图 2 DC-DE整合分析框架的工作流程 Fig. 2 Flowchart of the DC–DE integration analytical framework. Using the analysis of two cancer datasets as an example, we first identified tumor and normal co-expression modules for each dataset, then extracted the significantly conserved tumor/normal modules across individual datasets using pairwise Fisher's exact test (step 1). For each significantly conserved module, we performed integration-based DC (step 2) and DE (step 3) analyses to identify modules gaining co-expression, losing co-expression, up-regulated, and down-regulated in the tumor, respectively. Modules gaining co-expression and up-regulated in tumor were identified as activated modules and those losing co-expression and down-regulated in tumor were identified as dysfunctional modules. |
| |
首先利用R语言中GSCA程序包[15]识别肿瘤和癌旁样品之间显著的DC模块。对于一个包含n个基因的共表达模块M,计算其两两基因组成的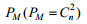
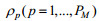


应用置换检验识别显著DC模块,置换检验的P值为
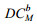
接着我们鉴定显著DC模块内的关键基因,对于一个包含n个基因的显著DC模块M,计算其中每个基因g的差异共表达分数DCg= 

首先基于Student’s t-test计算每组独立的微阵列表达谱中基因的差异表达P值,然后对T检验P值进行转换:当基因在肿瘤中上调时P值转化为pʹ=P/2,下调时转换为pʹ=1–P/2。因此当pʹ越接近0时,基因在肿瘤中越显著上调,越接近1时,基因在肿瘤中越显著下调。我们使用斯托弗法(Stouffer method) 合并两个独立微阵列表达谱中同一基因的转换P值为合并P值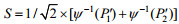

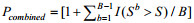
紧接着我们识别上下调的基因模块,对于一个包含n个基因的共表达模块M,首先计算其DE打分即
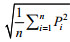
在DC和DE分析之后,我们发现显著DC模块内的基因相比非DC模块的基因具有更高的差异表达打分(DES),秩和检验表明两类模块内基因的差异表达打分具有显著差异(图 3)。这一现象表明在肿瘤中得到或失去相关性的DC基因也倾向于发生表达水平的变化,也佐证了DC和DE基因均与失调的生物学过程有关[2, 6-9, 32-33]。基于DC和DE的正相关关联,我们将同时满足DC和DE的基因模块定义为肿瘤相关模块(图 1底部)。肿瘤相关模块又进一步分为激活模块(显著DC且显著上调) 和失能模块(显著DC且显著下调)。在激活模块中,当一个基因是DC关键基因且其合并差异表达P值(Pcombined) < 0.05时,该基因为激活模块关键基因;在失能模块中,当一个基因是DC关键基因且其合并差异表达P值(Pcombined) > 0.95时,该基因为失能模块的关键基因。

|
| 图 3 DC模块基因与非DC模块基因的相关性比较 Fig. 3 Correlation between DC and DE genes. Tumor (A–C) and normal (D–F) co-expression modules of liver, stomach and colon cancer were respectively divided into significantly DC modules and the other ones. The combined DE scores of genes included in significantly DC modules are compared with those of the other modules through Wilcoxon rank-sum test and shown in box plots. The Wilcoxon rank-sum test P-values are shown above the box plots. |
| |
在得到肝癌、胃癌和结直肠癌各自的肿瘤相关模块和其中的关键基因后,我们将上述3种癌症的各两组微阵列数据作为训练集,将关键基因的表达量作为特征训练了针对3种癌症的随机森林模型。我们利用这些模型分析一系列测试集,以考察其区分肿瘤和癌旁样本的效能。测试集包括从GEO数据库中下载的与肝癌、胃癌和结直肠癌相关的3组微阵列数据GSE45436 (41个癌旁和62个肿瘤样品)、GSE7993 (10个癌旁和10个肿瘤样品) 和GSE18105 (17个癌旁和17个肿瘤样品),以及TCGA数据库下载的与肝癌(50例肿瘤和癌旁组织)、胃癌(27例肿瘤和癌旁组织) 和结直肠癌(41例肿瘤和癌旁组织) 相关的转录组测序(RNA Sequencing) 数据。
1.2.6 关键基因的Kaplan–Meier生存分析利用文本挖掘的方式,我们搜索了每种癌症的关键基因是否已被报道为癌症预后标志物。针对未报道的关键基因,我们进一步利用Kaplan- Meier生存曲线分析其对癌症存活率的影响。生存曲线使用了来自TCGA数据库的3种癌症的临床数据和RNA测序数据,分别包括363、348和430位肝癌、胃癌和结直肠癌患者。具体方法为以特定基因在患者中的表达量从大到小对患者排序,前1/4的患者为高表达组,后1/4的患者为低表达组,基于总体生存率(Overall survival rate) 绘制两类患者的生存曲线,当Log-rank检验P值小于0.05时认为两类患者的预后存在显著差异,该基因为癌症预后标志物。
1.2.7 肿瘤相关模块基因的通路富集分析我们使用R语言中程序包“ReactomePA”和“clusterProfiler”分别对激活和失能模块基因进行通路富集分析。两个软件包均使用双侧费舍尔精确检验,分别基于Reactome[34]和KEGG数据库识别显著通路,费舍尔精确检验P值小于0.05的通路具有统计学意义。
2 结果与分析 2.1 整合DC和DE的分析框架DC & DEmodule为了有效研究共表达模块的DC和DE特征,我们开发了一种新颖的整合DC和DE特征的分析框架DC & DEmodule并将其应用于肝癌、胃癌和结直肠癌的6个微阵列表达谱中,最终分别鉴定了2个(122个基因)、5个(108个基因) 和2个(23个基因) 激活模块,以及5个(144个基因)、5个(281个基因) 和1个(68个基因) 失能模块。详见补充表S3–S5。
为了考察肿瘤相关模块涉及的生物过程,我们分别基于KEGG和Reactome数据库进行了通路富集分析。针对肝癌、胃癌和结肠癌的激活模块,我们分别鉴定到21、44和27条KEGG通路以及187、418和214条Reactome通路。针对肝癌、胃癌和结肠癌的失能模块,我们鉴定到62、49和72条KEGG通路以及151、153和243条Reactome通路(详见补充表S6)。
我们首先考察了不同癌症共有和差异的通路(详见补充表S7)。我们发现22条KEGG通路和249条Reactome通路在至少两种癌症的激活模块中富集(图 4A和4C)。这些通路大多涉及细胞生长和凋亡,例如“DNA复制”,“细胞周期”和“p53信号传导通路”以及一些癌症相关的代谢过程,例如“嘧啶代谢”和“糖酵解/糖异生”等(图 4E和4G)。这些结果与目前学术界的认知是一致的,即所有癌症都呈现不受控制的细胞增殖以及核苷酸生物合成的上调[35]。其中,我们发现了一条Reactome通路“SLIT和ROBO1的表达调控(Regulation of expression of SLITs and ROBOs)”在3种癌症中均被富集到。先前也有研究表明SLIT2和ROBO1蛋白在多种癌症(例如乳腺癌、前列腺癌、卵巢癌、肺癌、肝癌和结肠癌) 中显著差异且被认为是多种致癌信号的重要调控因子[36-38]。在3种癌症的失能模块中,我们发现41条KEGG通路和89条Reactome通路在至少两种癌症中被富集(图 4B和4D)。这些通路多与代谢过程有关(图 4F和4H),包括氨基酸代谢(例如“氨基酸的生物合成” “组氨酸代谢” “色氨酸代谢”和“酪氨酸代谢”)、脂质代谢(例如“脂肪酸降解” “甾类激素生物合成”和“不饱和脂肪酸的生物合成”)、碳水化合物代谢(例如“柠檬酸循环”和“丙酮酸代谢”) 以及能量代谢(例如“硫代谢”和“氮代谢”)。也有研究表明,肿瘤中代谢相关基因表达呈总体下调趋势[35],与我们的研究一致。此外,我们还发现了一些癌症特异性的代谢通路,例如“胆汁分泌”、“胆汁酸和胆汁盐代谢”作为肝脏特有的生物过程仅在肝癌的失能模块中富集到[39]。总体而言,通路富集分析表明我们鉴定的肿瘤相关模块与关键的癌症通路息息相关。

|
| 图 4 通路富集分析总览 Fig. 4 Overview of pathway enrichment analysis. Venn diagrams of activated (A) and dysfunctional (B) KEGG pathways in liver, gastric and colon cancer. Venn diagrams of activated (C) and dysfunctional (D) Reactome pathways in liver, gastric and colon cancer. (E) Ten representative activated KEGG pathways shared by at least two cancers. (F) Ten representative dysfunctional KEGG pathways shared by at least two cancers. (G) Ten representative activated Reactome pathways shared by at least two cancers. (H) Ten representative dysfunctional Reactome pathways shared by at least two cancers. |
| |
为了评估DC & DEmodule框架的性能,我们将DC & DEmodule与现有的几种预测肿瘤相关通路的方法进行了比较。这些方法包括独立的DC分析(GSCA)、独立的DE分析(Student’s t-test)以及现有整合DC和DE分析的方法(DECODE)。我们利用以上方法分别处理3种癌症的微阵列表达谱,并对它们的结果进行KEGG和Reactome通路富集分析(详见补充表S8)。结果如下:独立的差异共表达分析方法即GSCA富集了37、42和54条被报道过的分别与3种癌症相关的KEGG通路和47、52和91条被报道过的分别与3种癌症相关的Reactome通路;独立的差异表达分析方法即T检验富集了17、15和28条被报道过的分别与三种癌症相关的KEGG通路以及100、112和30条被报道过的分别与3种癌症相关的Reactome通路;DECODE富集了41、48和49条被报道过的分别与3种癌症相关的KEGG通路以及97、117和118条被报道过的分别与3种癌症相关的Reactome通路;DC & DEmodule富集了83、93和99条被报道过的分别与3种癌症相关的KEGG通路以及338、571和457条被报道过的分别与3种癌症相关的Reactome通路(结果2.1部分)。
为了比较不同方法在通路预测上的效能,根据KEGG和Reactome数据库的通路注释,我们收集到与肝癌、胃癌和结直肠癌有关的58、57和57条KEGG通路以及190、187和188条Reactome通路(详见补充表S9)。将上述各类方法的通路富集结果与已知的癌症相关通路进行比较后发现,DC & DEmodule在鉴定通路总数及已知的癌症相关通路方面均具有最高的灵敏度(图 5A和5B)。

|
| 图 5 通路富集分析结果的比较 Fig. 5 Comparison of the pathway enrichment analysis. (A) The number of total KEGG pathways (line charts) and those reportedly cancer-related (bar charts) identified by different methods (DC–DE integration analytical framework, independent DC analysis, independent DE analysis, and DECODE) in liver, gastric and colon cancer. (B) The number of total Reactome pathways (line charts) and those reportedly cancer-related (bar charts) identified by different methods. |
| |
为了进一步探究模块内基因的功能,我们分别从肝癌、胃癌和结肠癌的激活模块中筛选了17、69和11个关键基因(详见材料与方法)。随后基于这些关键基因训练了3种癌症的随机森林分类器并对GEO和TCGA数据库中多组测试集的肿瘤和癌旁样本进行了分类(数据下载地址见补充表S10)。图 6A–C展示了各组数据分类结果的ROC曲线,曲线下的面积分别为0.99、0.91、0.96、0.90、0.88和0.95,其平均准确性达到93%,证明了关键基因与肿瘤和癌旁之间的差异密切相关,可以有效分离两者并且受不同来源数据的批次处理影响较小。

|
| 图 6 肿瘤相关模块和关键基因的评估 Fig. 6 Evaluation of the tumor-associated modules and hub genes. ROC curves of using the random forest classifiers trained by the liver (A), gastric (B) and colon (C) activated hub genes to classifier tumor and normal samples in additional datasets. (D) Overall survival plots of liver cancer patients in TCGA stratified by TCEB1 expression (log-rank P=0.002 5). (E) Overall survival plots of gastric cancer patients in TCGA stratified by RFC4 expression (log-rank P =0.032 6). (F) Overall survival plots of colon cancer patients in TCGA stratified by TRPC4AP expression (log-rank P=0.040 4). |
| |
为了研究关键基因与肿瘤预后的关系,我们对关键基因进行了生存分析。首先对鉴定到的17、69和11个关键基因进行文本挖掘。最终得到16、44和4个已报道与3种癌症相关的关键基因,其中包括15、35和3个已知的预后标志物(详见补充表S11)。对于未证明的关键基因,我们进行了Kaplan-Meier生存分析,发现了3个新的癌症预后生物标志物TCEB1、RFC4和TRPC4AP。第一,有学者发现TCEB1高表达时乳腺癌患者的预后较差[40],而我们发现TCEB1的高表达同样会导致肝癌患者的存活率降低(图 6D)。第二,以往研究发现RFC4高表达时宫颈癌患者的预后良好[41]但结肠癌和肝癌患者的预后较差[42-43]。在本研究中我们发现RFC4高表达提高了胃癌患者的生存率(图 6E)。第三,根据“人类蛋白质图谱(Human protein atlas)”,TRPC4AP高表达时肝癌和肾癌患者预后较差[44],而在这里我们发现TRPC4AP高表达提高了结直肠癌患者的生存率(图 6F)。经调研,3个新标志物均在癌症中具有潜在的调控作用。例如,TCEB1编码蛋白质Elongin C,它充当E3泛素连接酶复合体,介导HIF-1α降解,从而影响癌症中的血管生成和细胞增殖[45]。RFC4充当DNA聚合酶的引物识别因子,涉及“DNA复制”和“错配修复”通路[46]。TRPC4AP充当Myc特异性受体,通过将Myc桥接至E3连接酶复合物来促进Myc泛素化,Myc的降解影响细胞周期和肿瘤进展[47]。同一基因(例如RFC4和TRPC4AP) 往往在不同的癌症中具有相反的作用。与我们的发现一致,Li等发现RFC4可以根据肿瘤的细胞和组织学特征充当原癌基因或抑癌基因[48]。综上,癌症预后是个复杂多样的过程,所以对基因的精确调控机制还需要更严格的实验和评估。
3 讨论当前,尽管蛋白质质谱和单细胞测序等新兴的实验技术正在迅速发展,但传统技术产出的大量微阵列数据和RNA测序数据尚未得到充分挖掘。因此,亟需开发有效的数据分析方法对公共数据库中大批量转录组表达谱进行整合和二次分析。此外,基因间的相互作用关系复杂而多样,通过鉴定基因共表达模块来研究生物系统中基因间的关系,可以在分子水平上发现影响癌症和其他疾病发生发展的关键因素。在本研究中,我们提出了一种整合DC分析和DE分析同时整合不同来源数据集的分析框架DC & DEmodule,可以从疾病/健康表达谱中识别激活和功能失调的共表达基因模块。一系列评估和比较表明,DC & DEmodule框架可以有效发现肝癌、胃癌和结直肠癌中激活和失调的生物学通路和关键基因。
此外,针对3种癌症的关键基因的生存分析表明,同一基因在不同的癌症类型的预后中往往具有相反的作用。因此,本研究中整合同种癌症类型的多组数据集将有助于消除批次效应并帮助阐明不同癌症类型之间的真正差异。总而言之,我们相信新的分析框架可以整合公共数据库中快速累积的表达谱,帮助研究人员发现更多人类疾病中功能失调的生物过程和关键基因,为揭露复杂疾病的发生发展机制提供新的视角。
| [1] |
Liu ZP, Wang Y, Zhang XS, et al. Network-based analysis of complex diseases. IET Syst Biol, 2012, 6(1): 22-33. DOI:10.1049/iet-syb.2010.0052
|
| [2] |
Stuart JM, Segal E, Koller D, et al. A gene-coexpression network for global discovery of conserved genetic modules. Science, 2003, 302(5643): 249-255. DOI:10.1126/science.1087447
|
| [3] |
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics, 2008, 9(1): 1-13. DOI:10.1186/1471-2105-9-1
|
| [4] |
Oldham MC, Konopka G, Iwamoto K, et al. Functional organization of the transcriptome in human brain. Nature Neurosci, 2008, 11(11): 1271-1282. DOI:10.1038/nn.2207
|
| [5] |
Dewey FE, Perez MV, Wheeler MT, et al. Gene coexpression network topology of cardiac development, hypertrophy, and failure. Circ Cardiovasc Genet, 2011, 4(1): 26-35. DOI:10.1161/CIRCGENETICS.110.941757
|
| [6] |
Mitra K, Carvunis AR, Ramesh SK, et al. Integrative approaches for finding modular structure in biological networks. Nat Rev Genet, 2013, 14(10): 719-732. DOI:10.1038/nrg3552
|
| [7] |
Segal E, Shapira M, Regev A, et al. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet, 2003, 34(2): 166-176. DOI:10.1038/ng1165
|
| [8] |
Barabási AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet, 2011, 12(1): 56-68. DOI:10.1038/nrg2918
|
| [9] |
Barabasi AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet, 2004, 5(2): 101-113. DOI:10.1038/nrg1272
|
| [10] |
de la Fuente A. From 'differential expression' to 'differential networking'-identification of dysfunctional regulatory networks in diseases. Trends Genet, 2010, 26(7): 326-333. DOI:10.1016/j.tig.2010.05.001
|
| [11] |
Kostka D, Spang R. Finding disease specific alterations in the co-expression of genes. Bioinformatics, 2004, 20(suppl_1): i194-i199.
|
| [12] |
Carter SL, Brechbühler CM, Griffin M, et al. Gene co-expression network topology provides a framework for molecular characterization of cellular state. Bioinformatics, 2004, 20(14): 2242-2250. DOI:10.1093/bioinformatics/bth234
|
| [13] |
Hu R, Qiu X, Glazko G, et al. Detecting intergene correlation changes in microarray analysis: a new approach to gene selection. BMC Bioinformatics, 2009, 10(1): 1-9. DOI:10.1186/1471-2105-10-1
|
| [14] |
Mentzen WI, Floris M, de la Fuente A. Dissecting the dynamics of dysregulation of cellular processes in mouse mammary gland tumor. BMC Genomics, 2009, 10(1): 1-12. DOI:10.1186/1471-2164-10-1
|
| [15] |
Choi YJ, Kendziorski C. Statistical methods for gene set co-expression analysis. Bioinformatics, 2009, 25(21): 2780-2786. DOI:10.1093/bioinformatics/btp502
|
| [16] |
He D, Liu ZP, Honda M, et al. Coexpression network analysis in chronic hepatitis B and C hepatic lesions reveals distinct patterns of disease progression to hepatocellular carcinoma. J Mol Cell Biol, 2012, 4(3): 140-152. DOI:10.1093/jmcb/mjs011
|
| [17] |
Drag M, Skinkyté-Juskiené R, Do DN, et al. Differential expression and co-expression gene networks reveal candidate biomarkers of boar taint in non-castrated pigs. Sci Rep, 2017, 7(1): 1-18. DOI:10.1038/s41598-016-0028-x
|
| [18] |
Lui TW, Tsui NB, Chan LW, et al. DECODE: an integrated differential co-expression and differential expression analysis of gene expression data. BMC Bioinformatics, 2015, 16(1): 1-15. DOI:10.1186/s12859-014-0430-y
|
| [19] |
Mah WC, Thurnherr T, Chow PK, et al. Methylation profiles reveal distinct subgroup of hepatocellular carcinoma patients with poor prognosis. PLoS ONE, 2014, 9(8): e104158. DOI:10.1371/journal.pone.0104158
|
| [20] |
Yildiz G, Arslan-Ergul A, Bagislar S, et al. Genome-wide transcriptional reorganization associated with senescence-to-immortality switch during human hepatocellular carcinogenesis. PLoS ONE, 2013, 8(5): e64016. DOI:10.1371/journal.pone.0064016
|
| [21] |
D'Errico M, de Rinaldis E, Blasi MF, et al. Genome-wide expression profile of sporadic gastric cancers with microsatellite instability. Eur J Cancer, 2009, 45(3): 461-469. DOI:10.1016/j.ejca.2008.10.032
|
| [22] |
Wang Q, Wen YG, Li DP, et al. Upregulated INHBA expression is associated with poor survival in gastric cancer. Med Oncol, 2012, 29(1): 77-83. DOI:10.1007/s12032-010-9766-y
|
| [23] |
Uddin S, Ahmed M, Hussain A, et al. Genome-wide expression analysis of Middle Eastern colorectal cancer reveals FOXM1 as a novel target for cancer therapy. Am J Pathol, 2011, 178(2): 537-547. DOI:10.1016/j.ajpath.2010.10.020
|
| [24] |
Lin G, He X, Ji H, et al. Reproducibility Probability Score—incorporating measurement variability across laboratories for gene selection. Nat Biotechnol, 2006, 24(12): 1476-1477. DOI:10.1038/nbt1206-1476
|
| [25] |
Eschrich SA, Hoerter AM, Bloom GC, et al. Tissue-specific RMA models to incrementally normalize Affymetrix GeneChip data. Annu Int Conf IEEE Eng Med Biol Soc, 2008, 2419-2422.
|
| [26] |
Gaudet P, Argoud-Puy G, Cusin I, et al. neXtProt: organizing protein knowledge in the context of human proteome projects. J Proteome Res, 2013, 12(1): 293-298. DOI:10.1021/pr300830v
|
| [27] |
Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the dynamic tree cut package for R. Bioinformatics, 2008, 24(5): 719-720. DOI:10.1093/bioinformatics/btm563
|
| [28] |
Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA, 2003, 100(16): 9440-9445. DOI:10.1073/pnas.1530509100
|
| [29] |
Darlington RB, Hayes AF. Combining independent P values: extensions of the Stouffer and binomial methods. Psychol Methods, 2000, 5(4): 496-515. DOI:10.1037/1082-989X.5.4.496
|
| [30] |
Stouffer SA. A study of attitudes. Sci Am, 1949, 180(5): 11-15. DOI:10.1038/scientificamerican0549-11
|
| [31] |
Rhodes DR, Barrette TR, Rubin MA, et al. Meta-analysis of microarrays: interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer Res, 2002, 62(15): 4427-4433.
|
| [32] |
Hartwell LH, Hopfield JJ, Leibler S, et al. From molecular to modular cell biology. Nature, 1999, 402(6761): C47-C52.
|
| [33] |
Alon U. Biological networks: the tinkerer as an engineer. Science, 2003, 301(5641): 1866-1867. DOI:10.1126/science.1089072
|
| [34] |
Croft D, O'kelly G, Wu G, et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res, 2010, 39(suppl_1): D691-D697.
|
| [35] |
Hu J, Locasale JW, Bielas JH, et al. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat Biotechnol, 2013, 31(6): 522-529. DOI:10.1038/nbt.2530
|
| [36] |
Gara RK, Kumari S, Ganju A, et al. Slit/Robo pathway: a promising therapeutic target for cancer. Drug Discovery Today, 2015, 20(1): 156-164. DOI:10.1016/j.drudis.2014.09.008
|
| [37] |
Avci ME, Konu O, Yagci T. Quantification of SLIT-ROBO transcripts in hepatocellular carcinoma reveals two groups of genes with coordinate expression. BMC Cancer, 2008, 8(1): 1-11. DOI:10.1186/1471-2407-8-1
|
| [38] |
Huang T, Kang W, Cheng AS L, et al. The emerging role of Slit-Robo pathway in gastric and other gastro intestinal cancers. BMC Cancer, 2015, 15(1): 1-9. DOI:10.1186/1471-2407-15-1
|
| [39] |
Huang Q, Tan Y, Yin P, et al. Metabolic characterization of hepatocellular carcinoma using nontargeted tissue metabolomics. Cancer Res, 2013, 73(16): 4992-5002. DOI:10.1158/0008-5472.CAN-13-0308
|
| [40] |
Lindskog C. The Human Protein Atlas-an important resource for basic and clinical research. Expert Rev Proteomics, 2016, 13(7): 627-629. DOI:10.1080/14789450.2016.1199280
|
| [41] |
Tang X, Xu Y, Lu L, et al. Identification of key candidate genes and small molecule drugs in cervical cancer by bioinformatics strategy. Cancer Manag Res, 2018, 10: 3533. DOI:10.2147/CMAR.S171661
|
| [42] |
Xiang J, Fang L, Luo Y, et al. Levels of human replication factor C4, a clamp loader, correlate with tumor progression and predict the prognosis for colorectal cancer. J Transl Med, 2014, 12(1): 1-11. DOI:10.1186/1479-5876-12-1
|
| [43] |
Yang WX, Pan YY, You CG. CDK1, CCNB1, CDC20, BUB1, MAD2L1, MCM3, BUB1B, MCM2, and RFC4 may be potential therapeutic targets for hepatocellular carcinoma using integrated bioinformatic analysis. BioMed Res Int, 2019, 1245072.
|
| [44] |
Uhlén M, Fagerberg L, Hallström BM, et al. Tissue-based map of the human proteome. Science, 2015, 347(6220): 1260419. DOI:10.1126/science.1260419
|
| [45] |
Kamura T. Cullin-based E3 family. Tanpakushitsu Kakusan Koso, 2006, 51(10 Suppl): 1167-1172.
|
| [46] |
Ellison V, Stillman B. Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5' recessed DNA. PLoS Biol, 2003, 1(2): e33. DOI:10.1371/journal.pbio.0000033
|
| [47] |
Choi SH, Wright JB, Gerber SA, et al. Myc protein is stabilized by suppression of a novel E3 ligase complex in cancer cells. Genes Dev, 2010, 24(12): 1236-1241. DOI:10.1101/gad.1920310
|
| [48] |
Li Y, Gan S, Ren L, et al. Multifaceted regulation and functions of replication factor C family in human cancers. Am J Cancer Res, 2018, 8(8): 1343-1355.
|