 2022, Vol. 38
2022, Vol. 38




工业蛋白质理性设计与应用

http://dx.doi.org/10.13345/j.cjb.220586
中国科学院微生物研究所、中国微生物学会主办
中国科学院微生物研究所、中国微生物学会主办
文章信息
- 曲戈, 袁波, 孙周通
- QU Ge, YUAN Bo, SUN Zhoutong
- 工业蛋白质理性设计与应用
- Rational design and applications of industrial proteins
- 生物工程学报, 2022, 38(11): 4068-4080
- Chinese Journal of Biotechnology, 2022, 38(11): 4068-4080
- 10.13345/j.cjb.220586
-
文章历史
- Received: July 28, 2022
- Accepted: October 11, 2022
引用本文

|
曲戈, 袁波, 孙周通, 等. 工业蛋白质理性设计与应用. 生物工程学报, 2022, 38(11): 4068-4080.

QU G, YUAN B, SUN ZT, et al. Rational design and applications of industrial proteins. Chinese Journal of Biotechnology, 2022, 38(11): 4068-4080.
工业蛋白质理性设计与应用
1. 中国科学院天津工业生物技术研究所, 天津 300308;
2. 国家合成生物技术创新中心, 天津 300308
2. 国家合成生物技术创新中心, 天津 300308
摘要:作为合成生物学与绿色生物制造等领域的底层核心技术,蛋白理性设计可有效解决天然功能元件性能不足等共性挑战,创制高性能人工酶元件。值此天津工业生物研究所(Tianjin Institute of Industrial Biotechnology, TIB) 创立10周年之际,文中回顾了研究所在工业蛋白理性设计领域的系列重要工作进展。从酶设计方法学研究、新酶反应设计到生物催化应用等方面进行了分析讨论,并展望了本领域未来发展方向。望借此搭建学术界和产业界与酶理性设计的桥梁,促进新技术、新策略的开发应用,加速融合人工酶的基础研究与产业应用,推动我国生物制造领域的科技创新升级。
关键词:蛋白质工程 理性设计 催化机制 人工酶 生物催化
Rational design and applications of industrial proteins
1. Tianjin Institute of Industrial Biotechnology, Chinese Academy of Science, Tianjin 300308, China;
2. National Technology Innovation Center of Synthetic Biology, Tianjin 300308, China
2. National Technology Innovation Center of Synthetic Biology, Tianjin 300308, China
Abstract: As one of the underlying core technologies in the fields of synthetic biology and green bio-manufacturing, rational protein design is able to effectively solve generic challenges, e.g., improving insufficient performance of natural enzymes, and creating high-performance artificial enzymes. On the occasion of the 10th anniversary of the founding of the Tianjin Institute of Industrial Biotechnology (TIB), Chinese Academy of Sciences, this paper reviews the important progress of TIB achieved in rational design of industrial proteins, from the development of enzyme design methodology, the design of new enzyme reactions, to the applications of biocatalysis, and prospects future trends of this field. It is hoped that this will build a bridge between academia and industry on the rational design of enzymes, promote the development and application of new technologies and strategies. This will help merging the basic research and industrial application, thereby advancing the bio-manufacturing technological innovation.
Keywords:
protein engineering rational design catalytic mechanism artificial enzyme biocatalysis
工业酶作为生物制造产业的“芯片”,处于生物产业的核心地位并广泛应用于包括食品、饲料、纺织、材料、发酵、能源、精细化学品和化学药品制造等重要工业领域。2021年全世界酶制剂市场规模约70亿美元,并以4%年速率增长,支撑着其下游数十倍甚至数百倍的相关产业[1]。与国际先进水平相比,我国高性能生物催化剂无论在种类还是数量上均存在明显差距,导致目前产品成本高、品种数量少、产业竞争力不强等不利局面。因此,发展蛋白质工程改造技术对于我国生物产业的发展和经济模式的转变都具有特别重要的战略意义[2]。
蛋白质工程可追溯到20世纪70年代的定点突变技术(site-specific mutagenesis, SSM)[3]。其后50年的发展历程中涌现了一批改造方法与技术体系[4-6],并呈现先“两极分化”、后“合二为一”的发展趋势(图 1):其一是从一维序列入手(sequence-based),利用随机方式在编码蛋白质的DNA序列中引入突变,构建随机突变体文库,并经筛选等方式获得性能提升的蛋白质[7]。优点是只需蛋白质序列信息而不依赖其三维结构,典型的技术包括易错PCR (error-prone polymerase chain reaction, epPCR)[8]、DNA重组(DNA shuffling) 等[9];与此同时,另一条发展趋势基于蛋白质三维结构(structure-guided),结合酶催化机制,以理性的方式选取拟改造活性位点及构建单元,通过定点突变构建“小而精”的突变体文库来克服筛选瓶颈。其经典策略包括组合活性位点饱和突变(combinatorial active-site saturation test, CAST)[10]、迭代饱和突变(iterative saturation mutagenesis, ISM)[11-12]等。近年来随着计算技术的突飞猛进,数据驱动(data-driven) 的蛋白质计算设计逐渐融合了上述两种基于序列和结构的蛋白质改造策略的优点,经由先进算法指导的计算机虚拟筛选及从头设计,极大程度上缩减了蛋白质改造的工作周期[13-15]。

|
| 图 1 蛋白质工程发展之路 Fig. 1 Developmental of protein engineering. |
| |
过去10年,中国科学院天津工业生物技术研究所(以下简称“天津工业生物所”) 先后在“十二五”和“十三五”发展规划中将工业蛋白理性设计列为研究所重点培育方向之一,并率先牵头成立了工业酶国家工程实验室(后更名为工业酶国家工程研究中心),全面开展酶分子改造和工业酶应用等方向的关键技术研发。通过生物学与化学、晶体结构学、计算科学和工程学等学科的交叉与融合,解决生物催化剂活性低、工业环境下应用性能差等问题,实现医药中间体、天然食品香料、高值精细化学品和新型聚合物的高效生物合成,促进工业酶在化工、纺织、食品、制药、环境等领域中的应用,降低资源、能源消耗与污染物排放,促进节能减排与产业提升,为推动我国绿色生物制造研究和产业发展提供了有利支撑。下文将着重介绍天津工业生物所在酶设计方法开发、新酶设计应用等方面取得系列重要进展与突破。
1 创新工业酶理性设计技术体系如果将酶蛋白比喻为生物制造的“芯片”,先进的酶设计方法则被认为是保障芯片供给的“光刻机”。天津工业生物所聚焦高效、通用的酶设计新方法、新策略、新技术的研发,相继在精简密码子设计、拟突变位点的精准定位、聚焦突变技术,以及基于结构的系列设计策略等方面取得进展,为酶理性设计提供了新思路、新工具。
1.1 精简密码子设计与精准定位拟突变位点技术体系蛋白质的序列空间极为庞大,其所覆盖的可能突变体数目K的计算公式如公式(1) 所示[16],其中M代表所突变的位点数目;C为选用的氨基酸构建单元(密码子) 数目,例如采用20种天然氨基酸作为构建单元,则C即为20;X代表序列总长度。例如,当考虑一条由300个氨基酸组成的目标酶时,如果随机突变一个氨基酸位点,则产生约5 700个不同的突变体,如果同时发生两个位点突变,则有约1 600万个突变体,而如果是3个位点同时替换则会产生约300亿个突变体。从公式(1) 不难看出,蛋白质序列空间与两个因素直接相关:拟选用的密码子数目C,以及拟突变的位点数目M。因此,如何通过开发实用的关键技术,去掉冗余无效的密码子(降低C值),并理性定位要改造的氨基酸残基位点(减小M值),对于克服筛选瓶颈具有重要科学研究与应用价值。
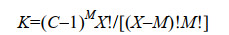
|
(1) |
目前常用的简并密码子有NNK (N代表4种碱基,K代表碱基A和C) 及NDT (D代表碱基A、C和T) 等,前者对应32个密码子,编码20种氨基酸;后者对应12个密码子,编码12种氨基酸。为有效降低C值,可基于酶催化机制解析、位点保守性分析、分子动力学模拟、Rosetta设计软件等技术,理性选取天然氨基酸种类(如极性/非极性、疏水/亲水、大位阻/小位阻等),进而精简拟选用的密码子数目,此谓之精简密码子设计。例如,笔者课题组以柠檬烯环氧水解酶和P450单加氧酶为研究对象,以理性设计的精简密码子(≤3种氨基酸) 为构建单元,并与传统NNK、NDT等多种简并密码子设计方法构建的突变体文库进行比较分析发现,在相同筛选工作量下,理性设计的精简密码子获得有益突变体数量显著增高,并基于统计学(Patrick/Firth及Nov计算) 分析得到理论支持[17]。这项工作不但证明了精简密码子设计与高覆盖度的筛选可加速有益突变体的获取,同时建立了精简密码子设计体系模型,揭示了密码子设计原则。
理性设计通常聚焦于酶催化口袋的活性位点,能否正确识别活性位点是减小M值的关键。基于酶催化过程的动态特征,以及酶结构自身的运动特性,笔者课题组证明了蛋白-配体相互作用指纹图谱技术[18-19]、构象动力学分析[20]、B-因子分析[21]等技术手段可用于高效定位活性位点。并在环氧水解酶底物特异识别[18]、羧酸还原酶催化活性提升[19, 22]、醇脱氢酶立体选择性改造[20, 23]等方面取得成功。通过这一系列工作,为关键氨基酸残基的精准定位奠定了技术基础,同时也为酶理性设计提供了理论基础。
1.2 理性聚焦迭代突变技术组合活性中心饱和突变/迭代饱和突变(Combinatorial active-site saturation test/Iterative saturation mutagenesis, CAST/ISM) 作为经常使用的半理性设计策略,尽管一定程度上克服了筛选瓶颈,但仍需建立“小而精”突变体文库,未完全摆脱对建库的依赖。2019年,吴起和Manfred T. Reetz教授等以南极假丝酵母脂肪酶B催化外消旋醇与外消旋酸的酯交换反应(双手性中心) 为模型,在活性位点引入体积大小差异的3种氨基酸(A/L/F) 及相似氨基酸作为构建单元,并经2−4轮酶分子进化,成功获得能够催化合成4种特异手性产物的系列突变体,且目标产物的立体选择性均 > 90%,并提出理性聚焦迭代突变技术(focused rational iterative site-specific mutagenesis, FRISM)[24]。随后,Manfred T. Reetz与孙周通研究员进一步开拓了FRISM技术体系,提出通过理性设计位点突变方案(图 2A),替代过去特定位点的饱和突变,进而避免突变体文库的构建[25],将定向进化与理性设计融为一体,是饱和突变这一传统定向进化模式的重要补充。

|
| 图 2 FRISM策略指导酶理性设计[25-26] Fig. 2 FRISM guided rational enzyme design[25-26]. 拟突变位点所对应氨基酸构建单元的理性选取[25] (A) 及其指导的糖基转移酶区域选择性改造案例[26] (B) Rationally chosen amino acids as building blocks for targeted residues[25] (A), and a case study of regioselectivity engineering towards a glucosyltransferase[26] (B). |
| |
黄酮类糖苷是一类具有多种生物活性的化合物。化学法合成特定糖苷反应需要多步的保护与去保护步骤,合成途径较为繁琐[27]。利用糖基转移酶进行糖基化反应,具有合成步骤少、反应条件温和等特点,但对于含有多羟基的黄酮类化合物,目前筛选到的糖基转移酶常获得不同比例的混合糖苷,如何控制糖基转移酶的区域选择性,获得单一糖苷产物极富挑战。孙媛霞团队与笔者团队合作,采用FRISM策略,融合序列保守性分析、晶体结构信息以及Rosetta酶设计等方法,对罗汉果来源糖基转移酶UGT74AC2进行理性设计(图 2B)。野生型糖基转移酶能够催化黄酮类化合物水飞蓟宾并生成3-O-葡萄糖苷、7-O-葡萄糖苷以及3, 7-O-双葡萄糖苷混合产物(产物比例22%︰39%︰39%)。利用理性聚焦设计策略,仅测试不到100个突变体,即成功获得能分别催化水飞蓟宾C3 (94%)、C7 (> 99%) 以及C3, 7位(> 99%) 羟基化的突变体,其对含氮和硫底物的催化功能也得以实现[26]。
同样采用FRISM策略,孙媛霞团队针对磷酸酶的底物特异性进行理性设计。通过分子对接、B-因子分析等方式理性选取关键氨基酸位点,并引入相应突变,增强酶与底物的氢键相互作用,获得了一系列对6-磷酸葡萄糖、6-磷酸果糖及6-磷酸甘露糖特异选择性催化的突变体。其特异性提高了12 000倍,成功实现了利用麦芽糊精和蔗糖合成甘露糖、果糖等产物[28]。此外,吴起团队利用FRISM策略对脂肪酸光脱羧酶的底物结合区域及活性区域进行突变位点设计,提升了对短链、中链、长链等多种脂肪族及芳香族羧酸的脱羧反应活性,拓展了光脱羧酶脱羧氘化反应底物特异性[29]。
上述工作证明了相比于传统饱和突变,FRISM策略更加聚焦位点及构建单元的理性选取,进而显著减少了筛选工作量,体现了FRISM策略指导理性设计的重要性。
1.3 镜像设计策略如果两个分子互为镜像对称,则称之为手性分子,许多天然产物及医药中间体均是手性分子。如何通过酶催化获得单一手性分子,是理性设计的重要研究方向。由于酶立体选择性偏好与其口袋几何构型有直接关联,特定的口袋几何构型决定了催化产物的手性。因此,通过“外科手术”式精准调控催化口袋活性位点,使其能够镜像识别潜手性底物,进而达到高立体选择性催化的目的同样充满挑战。
以腈水解酶催化合成手性氨基酸为例,尽管目前已有文献报道腈水解酶可催化3-取代戊二腈去对称化水解合成手性γ-氨基丁酸类化合物,然而天然腈水解酶催化3-烷基或3-芳基戊二腈生成(S)-和/或(R)-构型产物的对映体过量值较低[30-31]。在不损失酶活的基础上实现其立体选择性的翻转仍然是一大挑战。基于此,朱敦明、吴洽庆课题组通过对来源于集胞藻属(Synechocystis sp.) PCC6803的腈水解酶(SsNIT) 底物结合口袋进行镜像分析发现,两个影响立体选择性的关键氨基酸残基W170与V198处于互为镜像位置,其体积差异较大且分别与底物潜手性碳原子的两个取代基团有相互作用(图 3)。通过置换这两个氨基酸残基的位置,实现了该酶对3-异丁基戊二腈水解反应立体偏好性的翻转。进一步采用定点突变及组合突变技术重塑其底物结合口袋,获得了高立体选择性(> 99% ee) 及活性的突变体E8[32]。
这项工作证明了镜像设计策略可以很好地调控酶的立体选择性,并可用于腈水解酶及其他酶催化潜手性化合物去对称化反应的立体选择性改造,为获得立体偏好性互补的酶催化剂提供了一种新方法。
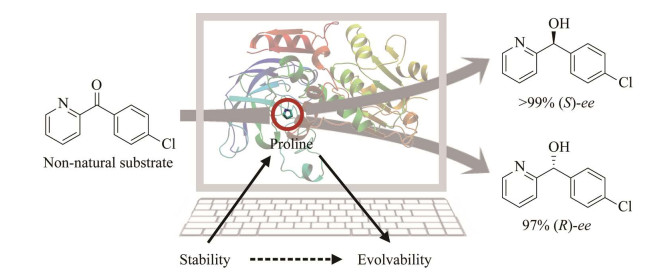
1.4 脯氨酸诱导设计策略随机突变与理性设计分别从序列和结构的角度出发,在酶工程领域都取得了巨大成功(图 1)。然而无论是序列还是结构,其本质都是进化的产物,而酶之所以能够被人为进化,是因为它具有一定程度的可进化性(evolvability);不同类型酶的改造难度并不一致,也从侧面反映了酶的可进化性不尽相同。然而如何精准定位酶蛋白关键氨基酸残基位点,通过位点突变解锁其可进化能力并赋予人工酶新颖的催化特性的问题,仍缺乏有效解决手段。
目前已知酶蛋白的可进化性与其结构稳定性有一定对应关系。在此基础上,笔者课题组开发了脯氨酸诱导环(loop) 区域工程测试(proline-induced loop engineering test, PiLoT) 的新酶设计方法。为验证PiLoT方法的可行性及优越性,选取醇脱氢酶TbSADH及其非天然潜手性酮底物为研究对象,通过PiLoT定位该酶催化口袋远端位点的第84位脯氨酸残基(P84) 为改造热点,在其结构稳定性可容忍的范围之内调控酶分子动力学(dynamics) 特性,进而提升酶分子的构象可塑性,有效地在酶催化口袋之外的区域引入构象变化。经计算机虚拟突变分析及后续迭代突变实验,获得了催化活性及立体选择性均大幅提升的组合突变体P84S/I86L及ΔP84/A85G,两者对应的ee值分别为 > 99% (S) 及97% (R) (图 4)。吉布斯自由能的变化(ΔΔG) 进一步表明突变体所增强的可进化能力是以降低稳定性的方式获得的。通过分子动力学模拟分析,揭示了P84位点突变诱导的催化口袋重塑是导致相关突变体立体选择性控制的分子基础[33]。
这项工作通过精准调控脯氨酸位点的稳定性与可进化性之间的平衡,即牺牲稳定性而使酶获得了可进化性,赋予其新的催化性能。由于脯氨酸的特殊刚性结构,蛋白质二级结构交界处较为频繁地使用脯氨酸。因此可以预见,PiLoT在指导其他酶改造中也将发挥重要作用。
2 新酶设计应用目前,天津工业生物所已搭建完成了较为成熟的酶设计平台,从上游的酶序列挖掘、催化机制解析,到计算设计,再到下游的实验反馈,不但能够快速获得高性能人工酶,同时还为其催化新反应应用铺平了道路。
2.1 医药中间体合成关键酶设计如何通过理性设计精准构筑药物分子中的多个手性中心,并获得单一构型的产物是酶设计领域的重要研究方向。左炔诺孕酮、孕二烯酮2018年零售药物销售额位列全球前200名内,其销售总额逾20亿美元,是两种非常重要的小分子避孕药物。(13R, 17S)-ethyl secol是合成这两种药物的关键中间体。该分子含有2个相同的羰基及1个季碳原子,因此可能有10种构型的还原产物(图 5A)。从20世纪60年代至今,科学界一直没有解决该分子合成过程中立体选择性差、转化效率低、产物复杂的难题。

|
| 图 5 野生型羰基还原酶及其突变体F12晶体结构[34] Fig. 5 Crystal structures of wild-type and mutant F12[34]. A:野生型酶与突变体结构比较;B:α6-helix局部结构;C:F12在α6-螺旋区域的突变位点示意图 (A) Superposition of the crystal structures of wild-type and mutant F12. (B) A magnification of the α6-helix region. (C) A detailed active site view of F12 α6-helix. |
| |
针对此挑战,朱敦明、吴洽庆课题组首先筛选评估了天津工业生物所酶库中的上百种羰基还原酶,选定来源于雷尔氏菌(Ralstonia sp.) 的羰基还原酶RasADH作为研究对象。利用结构指导的迭代设计策略,获得了优势突变体RasADH F12 (I91V/I187S/I188L/Q191N/F205A),该突变酶对模式底物的活力提高了183倍,目标产物(13R, 17S) 的比例由37.3%提高至 > 99.5%,双羰基还原产物比例由18.8%降低至 < 0.1%[34]。为阐明突变体F12性能提升的分子机制,作者对F12和该酶野生型晶体结构进行了解析和比较(图 5A)。发现尽管两者空间结构高度相似,然而在α6-螺旋区域存在较大差异(图 5B),突变体F12所包含的5个突变位点中,有4个位点(I91V, I187S, I188L, F205A) 均集中在此区域,通过提供相应的范德华力及疏水作用力等相互作用,促进了立体选择性催化合成(13R, 17S)-产物(图 5C)[34]。
这项工作为人工酶理性设计实现多手性复杂分子的精准构筑提供了理论基础,并且利用最优突变体实现了公斤级底物的转化,为工业化应用奠定了基础。此外,同样基于结构指导的设计策略,研究所还在人工酶催化合成手性氨基丁醇[35-36]、手性β-氨基酸[37]等方面取得系列研究进展,为酶法合成药物分子的手性砌块提供了新的思路。
2.2 一碳利用途径关键酶设计改造一碳利用途径关键酶,助力实现“双碳”目标,推动我国绿色生物制造升级,是工业酶理性设计的重要任务之一。
作为细胞生命中能量与物质代谢的枢纽,乙酰辅酶A是一碳利用途径的重要节点。天然生物合成乙酰辅酶A途径涉及8–10步以上的反应,且合成效率较为低下。江会锋团队利用新酶设计技术,从头设计了羟基乙醛合酶和乙酰磷酸合酶,并基于此构建了甲醛经3步反应到乙酰辅酶A合成的人工途径(synthetic acetyl-CoA pathway, SACA途径)[38]。人工设计的SACA途径突破了生物体固有代谢网络限制,具有化学驱动力强、不需要能量输入、与中心代谢正交和没有碳损失等优点,是首条乙酰辅酶A的人工生物合成途径,为生物一碳利用合成蛋白质、糖类、脂肪酸、天然产物等奠定了元件与途径基础。
此外,围绕一碳化合物甲醛的生物转化利用,朱蕾蕾课题组对关键碳链延长酶、甲醛裂合酶,进行了系统的分子改造并取得了突破性进展,大幅提升了其甲醛耐受性(1 mol/L甲醛) 并在此基础上设计构建了化学酶法转化甲醛为乳酸[39]和乙醇酸[40]的新转化路径,为一碳化合物的转化利用提供了新模式;大幅提升了甲醛裂合酶的底物亲和力(< 5 mmol/L甲醛) 和C3产物选择性,为“CO2人工合成淀粉”提供了高效人工酶元件[41]。
2.3 天然产物合成关键酶设计植物天然产物肉桂基-(6′-O-β-吡喃木糖基)-O-β-吡喃葡萄糖苷(肉桂醇甙E) 作为一种天然肉桂醇甙类似物,具有保护神经、护肝、抗癌、老年痴呆防治等功效,目前主要通过濒危药用植物玫瑰红景天提取,效率低下且破坏生态环境。刘涛团队与笔者团队合作,通过精准定位及理性设计长春花来源糖基转移酶CaUGT3的UDP-葡糖结合关键位点,成功将其改造为木糖基转移酶,并经组合突变进一步提升催化活力。随后将该突变体及其他异源基因导入宿主大肠杆菌体内,构建了肉桂醇甙E的人工细胞工厂,在5 L生物反应器中其产量达782.0 mg/L[42]。
氢化可的松作为自然界天然存在的糖皮质激素分子,具有抗病毒、消炎、抗过敏及抗休克等重要药理学作用,也可作为其他类固醇药物的合成前体。当前氢化可的松的工业化生产多采用化学法合成,常伴有副产物且得率较低。氢化可的松的生物合成途径中难度最大的一步,是控制对其C11位置的不对称羟化反应,仅生成β-羟基产物。然而天然P450羟化酶的立体选择性较差,可同时产生α-及β-羟基产物。张学礼团队与笔者团队合作,对蓝色犁头霉来源的P450羟化酶开展理性设计,成功将其催化β-立体选择性提升至86%;同时将该突变体导入酿酒酵母,并进行代谢工程改造,最终构建的酿酒酵母人工细胞生物转化合成氢化可的松的产量达1.06 g/L,是目前酵母体系生物转化合成氢化可的松的最高水平[43]。
2.4 抗生素母核合成途径关键酶设计作为一类重要的商业化抗生素药品,β-内酰胺类抗生素占据了全球抗生素市场一半以上的份额。而青霉素类和头孢类抗生素是两类主要的β-内酰胺类抗生素,这些抗生素母核的工业化生产主要依靠化学法或酶法,缺少在细胞工厂内从头合成的方法[44]。如果能在合成生物学常用底盘细胞,如酿酒酵母中实现β-内酰胺类母核的从头合成,对于抗生素的生产及研发新一代抗生素药物具有重要意义。
笔者团队通过人工酶设计与人工途径重构,获得人工酰化酶(图 6,红色箭头),实现了7-氨基头孢烷酸(7-amino cephalosporanic acid, 7-ACA) 的从头合成,产量超过6 μg/g干细胞重量(dry cell weight, DCW);随后,利用基因挖掘获得新酶元件,实现6-氨基青霉烷酸(6-amino penicillanic acid, 6-APA) 的从头合成,产量超过5 μg/g DCW;最后,创造性设计了人工扩环酶(图 6,蓝色箭头),实现了6-APA一步酶催化合成7-氨基去乙酰氧基头孢烷酸(7-amino desacetoxy cephalosporanic acid, 7-ADCA),产量约为1.7 μg/g DCW。基于上述人工酶,成功构建了酿酒酵母细胞工厂,实现了头孢菌素C、7-ACA、6-APA和7-ADCA在酿酒酵母中的从头合成,为酿酒酵母作为细胞工厂合成β-内酰胺类抗生素打下良好的基础[45]。
DNA合成是生命科学、生物医学、材料学及信息存储等众多领域的关键共性技术。当前DNA合成主要依赖化学合成法,其合成长度及成本存在难以逾越的极限,无法满足生物制造领域日益增长的需求。酶促DNA合成则在合成速度、长度、效率及成本等方面拥有化学合成无法比拟的潜力。因此,开发不依赖模板的高性能DNA聚合酶是解决下一代人工DNA合成技术的核心问题。
末端脱氧核苷酸转移酶(terminal deoxynucleotidyl transferase, TdT) 是常用的DNA聚合酶,然而天然TdT酶的底物结合口袋空间体积较小,对3′端带氧氨基保护的非天然核苷酸识别能力较差,无法满足合成需求[46]。为了实现寡核苷酸的高效可控合成,江会峰团队首先通过生物信息数据库的筛选与功能测试,获得来源于白喉雀的末端脱氧核苷酸转移酶ZaTdT,该酶对天然核苷酸底物初步展现出较好的催化活性。通过分子动力学模拟等手段,解析了该酶催化活性较已知同类酶大幅提升的分子机制,同时结合结构指导的酶理性设计技术,经过单点突变及组合突变,成功使该酶对修饰核苷酸底物的催化效率提升了3个数量级。改造后的人工酶可用于二步循环酶促DNA合成技术,合成DNA的平均准确率高达98.7%,与商业化的DNA化学合成法准确率相当[47]。
这项工作所开发的高性能人工DNA合成酶,不仅为核酸元件的设计再造提供有利工具,还有可能从根本上重塑DNA合成与编辑技术的发展面貌。
3 总结与展望本文探讨与总结了酶设计改造领域的发展历史与发展趋势,并回顾了天津工业生物所过去10年在酶理性设计领域所取得的重要进展。全面地开展酶理性设计方法学研究及新酶反应催化应用,对克服酶序列空间引发的筛选瓶颈,提出了多种解决方案,开辟了全新的酶设计应用场景。
当前人工智能(artificial intelligence, AI) 技术发展如火如荼,如何搭上AI技术的顺风车,加速酶理性设计与定向进化的融合,是蛋白质工程领域的重要发展趋势之一。在“十四五”期间,研究所继往开来,积极推动“人工酶的智能塑造技术”前沿探索。通过聚焦核心工业酶的设计原理等基本科学问题,研究蛋白功能与氨基酸序列进化关系,并开发由序列到功能的端对端一步设计策略和高度契合酶改造场景的人工智能新算法,快速构建高性能核心生物催化剂。为绿色生物制造提供核心元件支撑,同时为克服我国生物制造产业的底层技术供给瓶颈提供新思路。
参考文献
| [1] |
Tarafdar A, Sirohi R, Gaur VK, et al. Engineering interventions in enzyme production: lab to industrial scale. Bioresour Technol, 2021, 326: 124771. DOI:10.1016/j.biortech.2021.124771
|
| [2] |
曲戈, 朱彤, 蒋迎迎, 等. 蛋白质工程: 从定向进化到计算设计. 生物工程学报, 2019, 35(10): 1843-1856. Qu G, Zhu T, Jiang YY, et al. Protein engineering: from directed evolution to computational design. Chin J Biotech, 2019, 35(10): 1843-1856 (in Chinese). |
| [3] |
Hutchison CA 3rd, Phillips S, Edgell MH, et al. Mutagenesis at a specific position in a DNA sequence. J Biol Chem, 1978, 253(18): 6551-6560. DOI:10.1016/S0021-9258(19)46967-6
|
| [4] |
Wang YJ, Xue P, Cao MF, et al. Directed evolution: methodologies and applications. Chem Rev, 2021, 121(20): 12384-12444. DOI:10.1021/acs.chemrev.1c00260
|
| [5] |
Arnold FH. Innovation by evolution: bringing new chemistry to life (Nobel lecture). Angewandte Chemie Int Ed, 2019, 58(41): 14420-14426. DOI:10.1002/anie.201907729
|
| [6] |
Chen K, Arnold FH. Engineering new catalytic activities in enzymes. Nat Catal, 2020, 3(3): 203-213. DOI:10.1038/s41929-019-0385-5
|
| [7] |
曲戈, 赵晶, 郑平, 等. 定向进化技术的最新进展. 生物工程学报, 2018, 34(1): 1-11. Qu G, Zhao J, Zheng P, et al. Recent advances in directed evolution. Chin J Biotech, 2018, 34(1): 1-11 (in Chinese). |
| [8] |
Leung D, Chen E, Goeddel D. A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction. Technique, 1989, 1: 11-15.
|
| [9] |
Stemmer WPC. Rapid evolution of a protein in vitro by DNA shuffling. Nature, 1994, 370(6488): 389-391. DOI:10.1038/370389a0
|
| [10] |
Reetz MT, Bocola M, Carballeira JD, et al. Expanding the range of substrate acceptance of enzymes: combinatorial active-site saturation test. Angewandte Chemie Int Ed, 2005, 44(27): 4192-4196. DOI:10.1002/anie.200500767
|
| [11] |
Qu G, Sun ZT, Reetz M. Iterative saturation mutagenesis for semi-rational enzyme design. John Wiley & Sons, Ltd, 2021.
|
| [12] |
Zhao H, Lee S, Nielsen J, et al. Protein engineering: tools and applications. Wiley, 2021, 105-132.
|
| [13] |
卞佳豪, 杨广宇. 人工智能辅助的蛋白质工程. 合成生物学, 2022, 3(3): 429-444. Bian JH, Yang GY. Artificial intelligence-assisted protein engineering. Synth Biol J, 2022, 3(3): 429-444 (in Chinese). |
| [14] |
蒋迎迎, 曲戈, 孙周通. 机器学习助力酶定向进化. 生物学杂志, 2020, 37(4): 1-11. Jiang YY, Qu G, Sun ZT. Machine learning-assisted enzyme directed evolution. J Biol, 2020, 37(4): 1-11 (in Chinese). |
| [15] |
Bornscheuer UT, Huisman GW, Kazlauskas RJ, et al. Engineering the third wave of biocatalysis. Nature, 2012, 485(7397): 185-194. DOI:10.1038/nature11117
|
| [16] |
Reetz M. Directed evolution of selective enzymes: catalysts for organic chemistry and biotechnology. Wiley-VCH, 2016.
|
| [17] |
Li AT, Qu G, Sun ZT, et al. Statistical analysis of the benefits of focused saturation mutagenesis in directed evolution based on reduced amino acid alphabets. ACS Catal, 2019, 9(9): 7769-7778. DOI:10.1021/acscatal.9b02548
|
| [18] |
Sun ZT, Wu L, Bocola M, et al. Structural and computational insight into the catalytic mechanism of limonene epoxide hydrolase mutants in stereoselective transformations. J Am Chem Soc, 2018, 140(1): 310-318. DOI:10.1021/jacs.7b10278
|
| [19] |
Qu G, Liu BB, Zhang K, et al. Computer-assisted engineering of the catalytic activity of a carboxylic acid reductase. J Biotechnol, 2019, 306: 97-104. DOI:10.1016/j.jbiotec.2019.09.006
|
| [20] |
Liu BB, Qu G, Li JK, et al. Conformational dynamics-guided loop engineering of an alcohol dehydrogenase: capture, turnover and enantioselective transformation of difficult-to-reduce ketones. Adv Synth Catal, 2019, 361(13): 3182-3190. DOI:10.1002/adsc.201900249
|
| [21] |
Sun ZT, Liu Q, Qu G, et al. Utility of B-factors in protein science: interpreting rigidity, flexibility, and internal motion and engineering thermostability. Chem Rev, 2019, 119(3): 1626-1665. DOI:10.1021/acs.chemrev.8b00290
|
| [22] |
Qu G, Fu MX, Zhao LL, et al. Computational insights into the catalytic mechanism of bacterial carboxylic acid reductase. J Chem Inf Model, 2019, 59(2): 832-841. DOI:10.1021/acs.jcim.8b00763
|
| [23] |
Liu T, Bessembayeva L, Chen J, et al. Development of an economical fermentation platform for enhanced ansamitocin P-3 production in Actinosynnema pretiosum. Bioresour Bioprocess, 2019, 6(1): 1-10. DOI:10.1186/s40643-018-0235-3
|
| [24] |
Xu J, Cen Y, Singh W, et al. Stereodivergent protein engineering of a lipase to access all possible stereoisomers of chiral esters with two stereocenters. J Am Chem Soc, 2019, 141(19): 7934-7945. DOI:10.1021/jacs.9b02709
|
| [25] |
Qu G, Li AT, Acevedo-Rocha CG, et al. The crucial role of methodology development in directed evolution of selective enzymes. Angew Chem Int Ed Engl, 2020, 59(32): 13204-13231. DOI:10.1002/anie.201901491
|
| [26] |
Li J, Qu G, Shang N, et al. Near-perfect control of the regioselective glucosylation enabled by rational design of glycosyltransferases. Green Synth Catal, 2021, 2(1): 45-53. DOI:10.1016/j.gresc.2021.01.005
|
| [27] |
Chatterjee S, Moon S, Hentschel F, et al. An empirical understanding of the glycosylation reaction. J Am Chem Soc, 2018, 140(38): 11942-11953. DOI:10.1021/jacs.8b04525
|
| [28] |
Tian CY, Yang JG, Liu C, et al. Engineering substrate specificity of HAD phosphatases and multienzyme systems development for the thermodynamic-driven manufacturing sugars. Nat Commun, 2022, 13(1): 3582. DOI:10.1038/s41467-022-31371-8
|
| [29] |
Xu J, Fan JJ, Lou YJ, et al. Light-driven decarboxylative deuteration enabled by a divergently engineered photodecarboxylase. Nat Commun, 2021, 12(1): 3983. DOI:10.1038/s41467-021-24259-6
|
| [30] |
DeSantis G, Zhu ZL, Greenberg WA, et al. An enzyme library approach to biocatalysis: development of nitrilases for enantioselective production of carboxylic acid derivatives. J Am Chem Soc, 2002, 124(31): 9024-9025. DOI:10.1021/ja0259842
|
| [31] |
DeSantis G, Wong K, Farwell B, et al. Creation of a productive, highly enantioselective nitrilase through gene site saturation mutagenesis (GSSM). J Am Chem Soc, 2003, 125(38): 11476-11477. DOI:10.1021/ja035742h
|
| [32] |
Yu SS, Li JL, Yao PY, et al. Inverting the enantiopreference of nitrilase-catalyzed desymmetric hydrolysis of prochiral dinitriles by reshaping the binding pocket with a mirror-image strategy. Angew Chem Int Ed Engl, 2021, 60(7): 3679-3684. DOI:10.1002/anie.202012243
|
| [33] |
Qu G, Bi YX, Liu BB, et al. Unlocking the stereoselectivity and substrate acceptance of enzymes: proline-induced loop engineering test. Angew Chem Int Ed Engl, 2022, 61(1): e202110793.
|
| [34] |
Chen X, Zhang HL, Maria-Solano MA, et al. Efficient reductive desymmetrization of bulky 1, 3-cyclodiketones enabled by structure-guided directed evolution of a carbonyl reductase. Nat Catal, 2019, 2(10): 931-941.
|
| [35] |
Wang HY, Qu G, Li JK, et al. Data mining of amine dehydrogenases for the synthesis of enantiopure amino alcohols. Catal Sci Technol, 2020, 10(17): 5945-5952.
|
| [36] |
Tong FF, Qin ZM, Wang HY, et al. Biosynthesis of chiral amino alcohols via an engineered amine dehydrogenase in E. coli. coli. Front Bioeng Biotechnol, 2022, 9: 778584.
|
| [37] |
Zhang DL, Chen X, Zhang R, et al. Development of β-amino acid dehydrogenase for the synthesis of β-amino acids via reductive amination of β-keto acids. ACS Catal, 2015, 5(4): 2220-2224.
|
| [38] |
Lu XY, Liu YW, Yang YQ, et al. Constructing a synthetic pathway for acetyl-coenzyme A from one-carbon through enzyme design. Nat Commun, 2019, 10(1): 1378.
|
| [39] |
Li TZ, Tang ZJ, Wei HL, et al. Totally atom-economical synthesis of lactic acid from formaldehyde: combined bio-carboligation and chemo-rearrangement without the isolation of intermediates. Green Chem, 2020, 22(20): 6809-6814.
|
| [40] |
Li TZ, Tan ZJ, Tang ZJ, et al. One-pot chemoenzymatic synthesis of glycolic acid from formaldehyde. Green Chem, 2022, 24(13): 5064-5069.
|
| [41] |
Cai T, Sun HB, Qiao J, et al. Cell-free chemoenzymatic starch synthesis from carbon dioxide. Science, 2021, 373(6562): 1523-1527.
|
| [42] |
Bi HP, Qu G, Wang S, et al. Biosynthesis of a rosavin natural product in Escherichia coli by glycosyltransferase rational design and artificial pathway construction. Metab Eng, 2022, 69: 15-25.
|
| [43] |
Chen J, Fan FY, Qu G, et al. Identification of Absidia orchidis steroid 11β-hydroxylation system and its application in engineering Saccharomyces cerevisiae for one-step biotransformation to produce hydrocortisone. Metab Eng, 2020, 57: 31-42.
|
| [44] |
Hamed RB, Gomez-Castellanos JR, Henry L, et al. The enzymes of β-lactam biosynthesis. Nat Prod Rep, 2013, 30(1): 21-107.
|
| [45] |
Yang DM, Su WC, Jiang YY, et al. Biosynthesis of β-lactam nuclei in yeast. Metab Eng, 2022, 72: 56-65.
|
| [46] |
Loc'h J, Rosario S, Delarue M. Structural basis for a new templated activity by terminal deoxynucleotidyl transferase: implications for V(D)J recombination. Structure, 2016, 24(9): 1452-1463.
|
| [47] |
Lu XY, Li JL, Li CY, et al. Enzymatic DNA synthesis by engineering terminal deoxynucleotidyl transferase. ACS Catal, 2022, 12(5): 2988-2997.
|