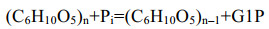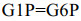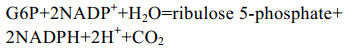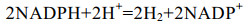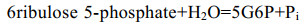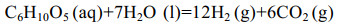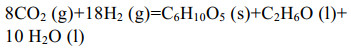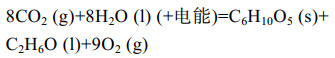| [1] |
Wang F, Harindintwali JD, Yuan ZZ, et al. Technologies and perspectives for achieving carbon neutrality. Innovation (Camb), 2021, 2(4): 100180.
|
|
| [2] | |
|
| [3] |
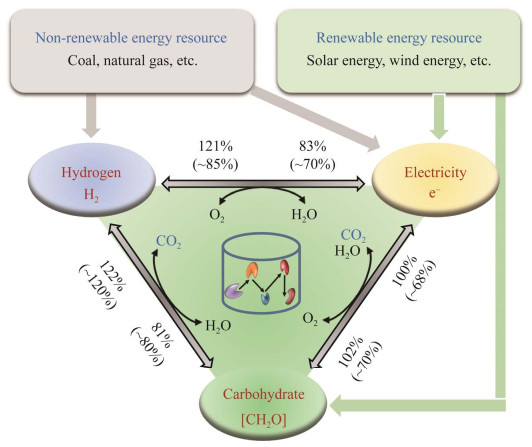
Zhang YHP, Huang WD. Constructing the electricity-carbohydrate-hydrogen cycle for a sustainability revolution. Trends Biotechnol, 2012, 30(6): 301-306. DOI:10.1016/j.tibtech.2012.02.006
|
|
| [4] |
Zhang YHP. A sweet out-of-the-box solution to the hydrogen economy: is the sugar-powered car science fiction?. Energy Environ Sci, 2009, 2(3): 272-282. DOI:10.1039/b818694d
|
|
| [5] | |
|
| [6] |
Liu JC, Wang SJ, Wei QS, et al. Present situation, problems and solutions of China's biomass power generation industry. Energy Policy, 2014, 70: 144-151. DOI:10.1016/j.enpol.2014.03.028
|
|
| [7] |
Huang WD, Percival Zhang YH. Energy efficiency analysis: biomass-to-wheel efficiency related with biofuels production, fuel distribution, and powertrain systems. PLoS One, 2011, 6(7): e22113. DOI:10.1371/journal.pone.0022113
|
|
| [8] |
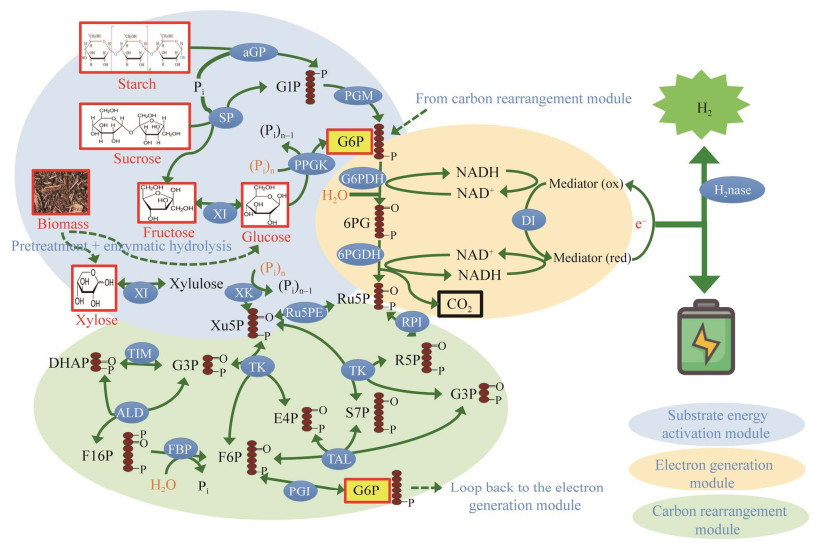
Zhang YHP. Production of biocommodities and bioelectricity by cell-free synthetic enzymatic pathway biotransformations: challenges and opportunities. Biotechnol Bioeng, 2010, 105(4): 663-677.
|
|
| [9] |
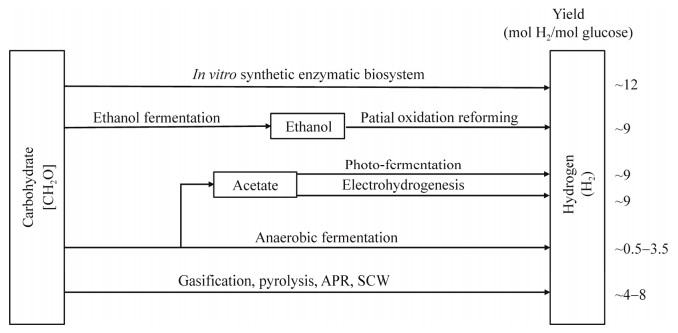
Zhang YHP, Sun JB, Zhong JJ. Biofuel production by in vitro synthetic enzymatic pathway biotransformation. Curr Opin Biotechnol, 2010, 21(5): 663-669. DOI:10.1016/j.copbio.2010.05.005
|
|
| [10] |
Zhu ZG, Tam TK, Percival Zhang YH. Cell-free biosystems in the production of electricity and bioenergy. Adv Biochem Eng Biotechnol, 2013, 137: 125-152.
|
|
| [11] |
Zhang YHP. Production of biofuels and biochemicals by in vitro synthetic biosystems: opportunities and challenges. Biotechnol Adv, 2015, 33(7): 1467-1483. DOI:10.1016/j.biotechadv.2014.10.009
|
|
| [12] |
You C, Shi T, Li YJ, et al. An in vitro synthetic biology platform for the industrial biomanufacturing of myo-inositol from starch. Biotechnol Bioeng, 2017, 114(8): 1855-1864. DOI:10.1002/bit.26314
|
|
| [13] |
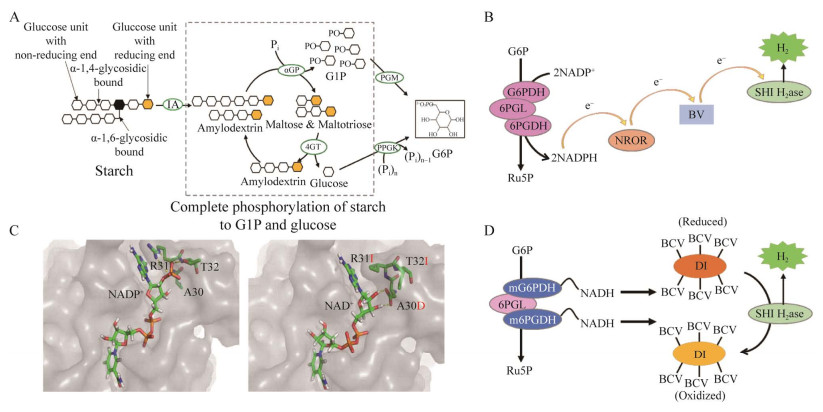
Kim EJ, Kim JE, Zhang YHPJ. Ultra-rapid rates of water splitting for biohydrogen gas production through in vitro artificial enzymatic pathways. Energy Environ Sci, 2018, 11(8): 2064-2072. DOI:10.1039/C8EE00774H
|
|
| [14] |
Zhu Z, Kin Tam T, Sun F, et al. A high-energy-density sugar biobattery based on a synthetic enzymatic pathway. Nat Commun, 2014, 5: 3026-3033. DOI:10.1038/ncomms4026
|
|
| [15] |
You C, Chen HG, Myung S, et al. Enzymatic transformation of nonfood biomass to starch. PNAS, 2013, 110(18): 7182-7187. DOI:10.1073/pnas.1302420110
|
|
| [16] |
Lim HJ, Kim DM. Cell-free synthesis of industrial chemicals and biofuels from carbon feedstocks. Curr Opin Biotechnol, 2022, 73: 158-163. DOI:10.1016/j.copbio.2021.08.002
|
|
| [17] |
Bowie JU, Sherkhanov S, Korman TP, et al. Synthetic biochemistry: the bio-inspired cell-free approach to commodity chemical production. Trends Biotechnol, 2020, 38(7): 766-778. DOI:10.1016/j.tibtech.2019.12.024
|
|
| [18] |
Taniguchi H, Okano K, Honda K. Modules for in vitro metabolic engineering: pathway assembly for bio-based production of value-added chemicals. Synth Syst Biotechnol, 2017, 2(2): 65-74. DOI:10.1016/j.synbio.2017.06.002
|
|
| [19] |
Ye XH, Wang YR, Hopkins RC, et al. Spontaneous high-yield production of hydrogen from cellulosic materials and water catalyzed by enzyme cocktails. ChemSusChem, 2009, 2(2): 149-52. DOI:10.1002/cssc.200900017
|
|
| [20] |
Del Campo JSM, Rollin J, Myung S, et al. High-yield production of dihydrogen from xylose by using a synthetic enzyme cascade in a cell-free system. Angew Chem Int Ed Engl, 2013, 52(17): 4587-4590. DOI:10.1002/anie.201300766
|
|
| [21] |
Myung S, Rollin J, You C, et al. In vitro metabolic engineering of hydrogen production at theoretical yield from sucrose. Metab Eng, 2014, 24: 70-77. DOI:10.1016/j.ymben.2014.05.006
|
|
| [22] |
Rollin JA, Martin Del Campo J, Myung S, et al. High-yield hydrogen production from biomass by in vitro metabolic engineering: mixed sugars coutilization and kinetic modeling. PNAS, 2015, 112(16): 4964-4969. DOI:10.1073/pnas.1417719112
|
|
| [23] |
Kim JE, Kim EJ, Chen H, et al. Advanced water splitting for green hydrogen gas production through complete oxidation of starch by in vitro metabolic engineering. Metab Eng, 2017, 44: 246-252. DOI:10.1016/j.ymben.2017.09.015
|
|
| [24] |
Huffman MA, Fryszkowska A, Alvizo O, et al. Design of an i n vitro biocatalytic cascade for the manufacture of islatravir. Science, 2019, 366(6470): 1255-1259. DOI:10.1126/science.aay8484
|
|
| [25] |
Cai T, Sun HB, Qiao J, et al. Cell-free chemoenzymatic starch synthesis from carbon dioxide. Science, 2021, 373(6562): 1523-1527. DOI:10.1126/science.abh4049
|
|
| [26] |
Rogers TA, Bommarius AS. Utilizing simple biochemical measurements to predict lifetime output of biocatalysts in continuous isothermal processes. Chem Eng Sci, 2010, 65(6): 2118-2124. DOI:10.1016/j.ces.2009.12.005
|
|
| [27] |
Myung S, Wang YR, Zhang YHP. Fructose-1, 6-bisphosphatase from a hyper-thermophilic bacterium Thermotoga maritima: characterization, metabolite stability, and its implications. Process Biochem, 2010, 45(12): 1882-1887. DOI:10.1016/j.procbio.2010.03.017
|
|
| [28] |
Wang YR, Percival Zhang YH. Overexpression and simple purification of the Thermotoga maritima 6-phosphogluconate dehydrogenase in Escherichia coli and its application for NADPH regeneration. Microb Cell Fact, 2009, 8: 30. DOI:10.1186/1475-2859-8-30
|
|
| [29] |
Giver L, Gershenson A, Freskgard PO, et al. Directed evolution of a thermostable esterase. PNAS, 1998, 95(22): 12809-12813. DOI:10.1073/pnas.95.22.12809
|
|
| [30] |
Zhang YHP, Myung S, You C, et al. Toward low-cost biomanufacturing through in vitro synthetic biology: bottom-up design. J Mater Chem, 2011, 21(47): 18877-18886. DOI:10.1039/c1jm12078f
|
|
| [31] |
Ye J, Li YJ, Bai YQ, et al. A facile and robust T7-promoter-based high-expression of heterologous proteins in Bacillus subtilis. Bioresour Bioprocess, 2022, 9(1): 1-12. DOI:10.1186/s40643-021-00489-w
|
|
| [32] | |
|
| [33] |
Nowak C, Pick A, Lommes P, et al. Enzymatic reduction of nicotinamide biomimetic cofactors using an engineered glucose dehydrogenase: providing a regeneration system for artificial cofactors. ACS Catal, 2017, 7(8): 5202-5208. DOI:10.1021/acscatal.7b00721
|
|
| [34] |
Wang W, Liu MX, You C, et al. ATP-free biosynthesis of a high-energy phosphate metabolite fructose 1, 6-diphosphate by in vitro metabolic engineering. Metab Eng, 2017, 42: 168-174. DOI:10.1016/j.ymben.2017.06.006
|
|
| [35] |
Meng DD, Wei XL, Zhang Y, et al. Stoichiometric conversion of cellulosic biomass by in vitro synthetic enzymatic biosystems for biomanufacturing. ACS Catal, 2018, 8(10): 9550-9559. DOI:10.1021/acscatal.8b02473
|
|
| [36] |
Wang YR, Huang WD, Sathitsuksanoh N, et al. Biohydrogenation from biomass sugar mediated by in vitro synthetic enzymatic pathways. Chem Biol, 2011, 18(3): 372-380. DOI:10.1016/j.chembiol.2010.12.019
|
|
| [37] |
Opgenorth PH, Korman TP, Bowie JU. A synthetic biochemistry module for production of bio-based chemicals from glucose. Nat Chem Biol, 2016, 12(6): 393-395. DOI:10.1038/nchembio.2062
|
|
| [38] | |
|
| [39] |
Wu YR, Zhang MM, Zhong MQ, et al. Synergistic enzymatic saccharification and fermentation of agar for biohydrogen production. Bioresour Technol, 2017, 241: 369-373. DOI:10.1016/j.biortech.2017.05.117
|
|
| [40] |
Veit A, Akhtar MK, Mizutani T, et al. Constructing and testing the thermodynamic limits of synthetic NAD(P)H: H 2 pathways. Microb Biotechnol, 2008, 1(5): 382-394. DOI:10.1111/j.1751-7915.2008.00033.x
|
|
| [41] |
Chou CJ, Jenney FE Jr, Adams MWW, et al. Hydrogenesis in hyperthermophilic microorganisms: implications for biofuels. Metab Eng, 2008, 10(6): 394-404. DOI:10.1016/j.ymben.2008.06.007
|
|
| [42] |
Zhang YHP, Evans BR, Mielenz JR, et al. High-yield hydrogen production from starch and water by a synthetic enzymatic pathway. PLoS One, 2007, 2(5): e456. DOI:10.1371/journal.pone.0000456
|
|
| [43] |
Moustafa H, Kim EJ, Zhu ZG, et al. Water splitting for high-yield hydrogen production energized by biomass xylooligosaccharides catalyzed by an enzyme cocktail. Chemcatchem, 2016, 8(18): 2898-2902. DOI:10.1002/cctc.201600772
|
|
| [44] |
Liao HH, Myung S, Zhang YHP. One-step purification and immobilization of thermophilic polyphosphate glucokinase from Thermobifida fusca YX: glucose-6-phosphate generation without ATP. Appl Microbiol Biotechnol, 2012, 93(3): 1109-1117. DOI:10.1007/s00253-011-3458-1
|
|
| [45] |
Kim EJ, Wu CH, Adams MWW, et al. Exceptionally high rates of biological hydrogen production by biomimetic in vitro synthetic enzymatic pathways. Chemistry, 2016, 22(45): 16047-16051. DOI:10.1002/chem.201604197
|
|
| [46] |
Huang R, Chen H, Zhong C, et al. High-throughput screening of coenzyme preference change of thermophilic 6-phosphogluconate dehydrogenase from NADP + to NAD +. Sci Rep, 2016, 6: 32644. DOI:10.1038/srep32644
|
|
| [47] |
Chen H, Zhu Z, Huang R, et al. Coenzyme engineering of a hyperthermophilic 6-phosphogluconate dehydrogenase from NADP + to NAD + with its application to biobatteries. Sci Rep, 2016, 6: 36311. DOI:10.1038/srep36311
|
|
| [48] |
Sakai H, Nakagawa T, Tokita Y, et al. A high-power glucose/oxygen biofuel cell operating under quiescent conditions. Energy Environ Sci, 2009, 2(1): 133-138. DOI:10.1039/B809841G
|
|
| [49] |
Cosnier S, Shan D, Ding SN. An easy compartment-less biofuel cell construction based on the physical co-inclusion of enzyme and mediator redox within pressed graphite discs. Electrochem Commun, 2010, 12(2): 266-269. DOI:10.1016/j.elecom.2009.12.011
|
|
| [50] |
Xu S, Minteer S. Enzymatic biofuel cell for oxidation of glucose to CO 2. ACS Catal, 2012, 2(1): 91-94. DOI:10.1021/cs200523s
|
|
| [51] |
Zhu ZG, Sun FF, Zhang XZ, et al. Deep oxidation of glucose in enzymatic fuel cells through a synthetic enzymatic pathway containing a cascade of two thermostable dehydrogenases. Biosens Bioelectron, 2012, 36(1): 110-115. DOI:10.1016/j.bios.2012.04.001
|
|
| [52] |
Zhu ZG, Wang YR, Minteer SD, et al. Maltodextrin-powered enzymatic fuel cell through a non-natural enzymatic pathway. J Power Sources, 2011, 196(18): 7505-7509. DOI:10.1016/j.jpowsour.2011.04.038
|
|
| [53] |
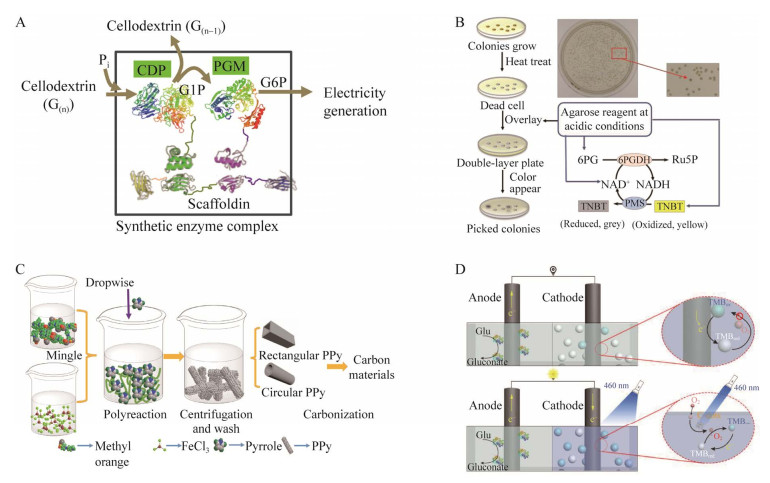
Meng DD, Wu RR, Wang J, et al. Acceleration of cellodextrin phosphorolysis for bioelectricity generation from cellulosic biomass by integrating a synthetic two-enzyme complex into an in vitro synthetic enzymatic biosystem. Biotechnol Biofuels, 2019, 12: 267. DOI:10.1186/s13068-019-1607-4
|
|
| [54] |
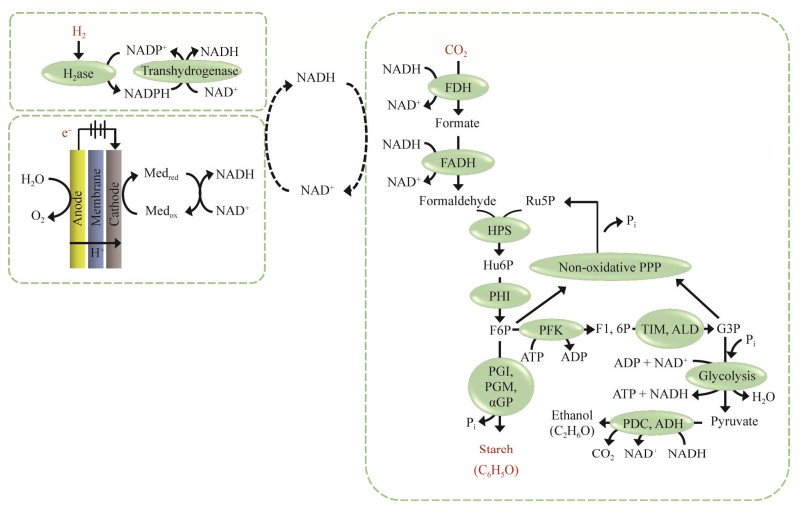
Zhu ZG, Zhang YHP. In vitro metabolic engineering of bioelectricity generation by the complete oxidation of glucose. Metab Eng, 2017, 39: 110-116. DOI:10.1016/j.ymben.2016.11.002
|
|
| [55] |
Zhu ZG, Ma CL, Zhang YHP. Co-utilization of mixed sugars in an enzymatic fuel cell based on an in vitro enzymatic pathway. Electrochimica Acta, 2018, 263: 184-191. DOI:10.1016/j.electacta.2017.11.083
|
|
| [56] |
Li GW, Wei XL, Wu RR, et al. Stoichiometric conversion of maltose for biomanufacturing by in vitro synthetic enzymatic biosystems. Biodesign Res, 2022, 2022: 1-11.
|
|
| [57] |
Wu RR, Ma CL, Zhang YHP, et al. Complete oxidation of xylose for bioelectricity generation by reconstructing a bacterial xylose utilization pathway in vitro. ChemCatChem, 2018, 10(9): 2030-2035. DOI:10.1002/cctc.201702018
|
|
| [58] |
Shi PK, Wu RR, Wang J, et al. Biomass sugar-powered enzymatic fuel cells based on a synthetic enzymatic pathway. Bioelectrochemistry, 2022, 144: 108008. DOI:10.1016/j.bioelechem.2021.108008
|
|
| [59] |
Wang LL, Gong WC, Wang F, et al. Efficient bienzyme nanocomposite film for chiral recognition of L-tryptophan, L-phenylalanine and L-tyrosine. Anal Methods, 2016, 8(17): 3481-3487. DOI:10.1039/C5AY03290C
|
|
| [60] |
Vargas E, Ruiz MA, Ferrero FJ, et al. Automatic bionalyzer using an integrated amperometric biosensor for the determination of L-malic acid in wines. Talanta, 2016, 158: 6-13. DOI:10.1016/j.talanta.2016.05.050
|
|
| [61] |
Sakamoto H, Komatsu T, Yamasaki K, et al. Design of a multi-enzyme reaction on an electrode surface for an L-glutamate biofuel anode. Biotechnol Lett, 2017, 39(2): 235-240. DOI:10.1007/s10529-016-2237-6
|
|
| [62] |
Hirano Y, Ikegami M, Kowata K, et al. Bienzyme reactions on cross-linked DNA scaffolds for electrochemical analysis. Bioelectrochemistry, 2017, 113: 15-19. DOI:10.1016/j.bioelechem.2016.08.005
|
|
| [63] |
Moehlenbrock MJ, Meredith MT, Minteer SD. Bioelectrocatalytic oxidation of glucose in CNT impregnated hydrogels: advantages of synthetic enzymatic metabolon formation. ACS Catal, 2012, 2(1): 17-25. DOI:10.1021/cs200482v
|
|
| [64] |
Behrendorff JBYH, Borràs-Gas G, Pribil M. Synthetic protein scaffolding at biological membranes. Trends Biotechnol, 2020, 38(4): 432-446. DOI:10.1016/j.tibtech.2019.10.009
|
|
| [65] |
Ma CL, Wu RR, Huang R, et al. Directed evolution of a 6-phosphogluconate dehydrogenase for operating an enzymatic fuel cell at lowered anodic pHs. J Electroanal Chem, 2019, 851: 113444. DOI:10.1016/j.jelechem.2019.113444
|
|
| [66] |
Kang ZP, Zhang YHP, Zhu ZG. A shriveled rectangular carbon tube with the concave surface for high-performance enzymatic glucose/O 2 biofuel cells. Biosens Bioelectron, 2019, 132: 76-83. DOI:10.1016/j.bios.2019.02.044
|
|
| [67] |
Li ZH, Kang ZP, Zhu ZG. A photo-switch for enzymatic biofuel cells based on the photo-oxidization of electron acceptor in cathode by C-dots nanozyme. Chem Eng J, 2022, 428: 131258. DOI:10.1016/j.cej.2021.131258
|
|
| [68] |
Song HY, Ma CL, Liu P, et al. A hybrid CO 2 electroreduction system mediated by enzyme-cofactor conjugates coupled with Cu nanoparticle-catalyzed cofactor regeneration. J CO2 Util, 2019, 34: 568-575. DOI:10.1016/j.jcou.2019.08.007
|
|
| [69] |
Song HY, Ma CL, Zhou W, et al. Construction of enzyme-cofactor/mediator conjugates for enhanced in vitro bioelectricity generation. Bioconjug Chem, 2018, 29(12): 3993-3998. DOI:10.1021/acs.bioconjchem.8b00766
|
|
| [70] | |
|
| [71] |
Wang YM, Song YH, Ma CL, et al. Electrochemical characterization of a truncated hydrogenase from Pyrococcus furiosus. Electrochimica Acta, 2021, 387: 138502. DOI:10.1016/j.electacta.2021.138502
|
|
| [72] |
Güven G, Prodanovic R, Schwaneberg U. Protein engineering-an option for enzymatic biofuel cell design. Electroanalysis, 2010, 22(7/8): 765-775.
|
|
| [73] |
Ma CL, Liu MX, You C, et al. Engineering a diaphorase via directed evolution for enzymatic biofuel cell application. Bioresour Bioprocess, 2020, 7(1): 1-11. DOI:10.1186/s40643-019-0289-x
|
|
| [74] |
Kang ZP, Wang YM, Yang CN, et al. Multifunctional N and O co-doped 3D carbon aerogel as a monolithic electrode for either enzyme immobilization, oxygen reduction and showing supercapacitance. Electrochimica Acta, 2021, 395: 139179. DOI:10.1016/j.electacta.2021.139179
|
|
| [75] |
Wang YM, Kang ZP, Zhang LL, et al. Elucidating the interactions between a[NiFe]-hydrogenase and carbon electrodes for enhanced bioelectrocatalysis. ACS Catal, 2022, 12(2): 1415-1427. DOI:10.1021/acscatal.1c05306
|
|
| [76] |
Li ZH, Kang ZP, Wu B, et al. A MXene-based slurry bioanode with potential application in implantable enzymatic biofuel cells. J Power Sources, 2021, 506: 230206. DOI:10.1016/j.jpowsour.2021.230206
|
|
| [77] |
Yang CN, Li ZH, Ma CL, et al. Photoswitchable enzymatic biofuel cell based on fusion protein with natural photoreceptor vivid. ACS Appl Bio Mater, 2022, 5(2): 459-464. DOI:10.1021/acsabm.1c01268
|
|
| [78] |
Mariano RG, McKelvey K, White HS, et al. Selective increase in CO 2 electroreduction activity at grain-boundary surface terminations. Science, 2017, 358(6367): 1187-1192. DOI:10.1126/science.aao3691
|
|
| [79] |
Umeda M, Niitsuma Y, Horikawa T, et al. Electrochemical reduction of CO 2 to methane on platinum catalysts without overpotentials: strategies for improving conversion efficiency. ACS Appl Energy Mater, 2020, 3(1): 1119-1127. DOI:10.1021/acsaem.9b02178
|
|
| [80] |
Wang JJ, Li GN, Li ZL, et al. A highly selective and stable ZnO-ZrO 2 solid solution catalyst for CO 2 hydrogenation to methanol. Sci Adv, 2017, 3(10): e1701290. DOI:10.1126/sciadv.1701290
|
|
| [81] |
Wu Y, Jiang Z, Lu X, et al. Domino electroreduction of CO 2 to methanol on a molecular catalyst. Nature, 2019, 575(7784): 639-642. DOI:10.1038/s41586-019-1760-8
|
|
| [82] |
Li J, Kuang Y, Meng Y, et al. Electroreduction of CO 2 to formate on a copper-based electrocatalyst at high pressures with high energy conversion efficiency. J Am Chem Soc, 2020, 142(16): 7276-7282. DOI:10.1021/jacs.0c00122
|
|
| [83] |
Duan YX, Zhou YT, Yu Z, et al. Boosting production of HCOOH from CO 2 electroreduction via Bi/CeO x. Angew Chem Int Ed, 2021, 60(16): 8798-8802. DOI:10.1002/anie.202015713
|
|
| [84] |
Huang Y, Mao XN, Yuan GT, et al. Size-dependent selectivity of electrochemical CO 2 reduction on converted In 2O 3 nanocrystals. Angew Chem Int Ed Engl, 2021, 60(29): 15844-15848. DOI:10.1002/anie.202105256
|
|
| [85] |
Song Y, Peng R, Hensley DK, et al. High-selectivity electrochemical conversion of CO2 to ethanol using a copper nanoparticle/N-doped graphene electrode. Chemistry Select, 2016, 1(19): 6055-6061.
|
|
| [86] |
Zhang YHP. Simpler is better: high-yield and potential low-cost biofuels production through cell-free synthetic pathway biotransformation (S yPaB). ACS Catal, 2011, 1(9): 998-1009. DOI:10.1021/cs200218f
|
|
| [87] |
Zhang YHP, You C, Chen H, et al. Surpassing Photosynthesis: High-efficiency and scalable CO2 utilization through artificial photosynthesis//Attalla. In recent advances in post-combustion CO2 capture chemistry. Washington, DC: American Chemical Society, ACS Symposium Series, 2012, 1097: 275-292.
|
|
 2022, Vol. 38
2022, Vol. 38