 2022, Vol. 38
2022, Vol. 38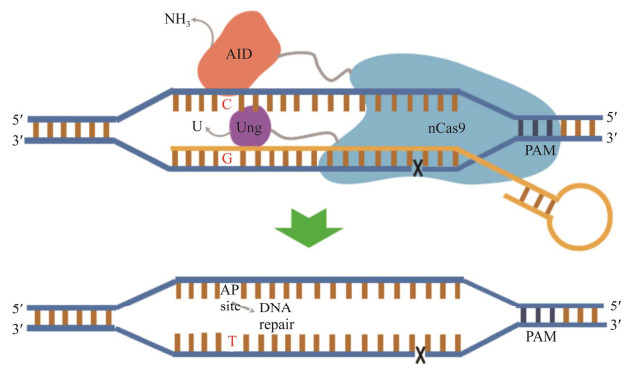



基因组编辑技术在工业生物领域中的应用现状及展望

http://dx.doi.org/10.13345/j.cjb.220566
中国科学院微生物研究所、中国微生物学会主办
中国科学院微生物研究所、中国微生物学会主办
文章信息
- 杨超, 董兴啸, 张学礼, 毕昌昊
- YANG Chao, DONG Xingxiao, ZHANG Xueli, BI Changhao
- 基因组编辑技术在工业生物领域中的应用现状及展望
- Application of genome editing technology in industrial microorganisms: current status and perspectives
- 生物工程学报, 2022, 38(11): 4132-4145
- Chinese Journal of Biotechnology, 2022, 38(11): 4132-4145
- 10.13345/j.cjb.220566
-
文章历史
- Received: July 21, 2022
- Accepted: October 27, 2022
引用本文

|
杨超, 董兴啸, 张学礼, 等. 基因组编辑技术在工业生物领域中的应用现状及展望. 生物工程学报, 2022, 38(11): 4132-4145.

YANG C, DONG XX, ZHANG XL, et al. Application of genome editing technology in industrial microorganisms: current status and perspectives. Chinese Journal of Biotechnology, 2022, 38(11): 4132-4145.
基因组编辑技术在工业生物领域中的应用现状及展望
1. 中国科学院天津工业生物技术研究所,天津 300308;
2. 大连工业大学 生物工程学院,辽宁 大连 116034
2. 大连工业大学 生物工程学院,辽宁 大连 116034
摘要:精准且高效的操纵基因表达或改写基因组序列是基因组编辑领域的研究热点,也是助力工业生物技术快速发展的核心使能技术。基因组编辑技术经历了从锌指核酸酶(zinc finger nucleases, ZFNs)、转录激活因子样效应物核酸酶(transcription activator-like effector nucleases, TALEN) 和Cas核酸酶3个发展阶段,目前随着CRISPR/Cas这一基因组编辑工具的蓬勃发展,研究人员在工业生物中建立了一系列基于该技术的一代及二代Cas基因组编辑技术,助力大肠杆菌等原核及酿酒酵母等真核工业生物底盘细胞的建立及优化。本文对目前基于常用底盘细胞的工业生物技术进行介绍,并对技术的发展历程、应用效果等特点进行总结,最后展望工业生物技术的未来发展前景,以期为科研人员提供参考。
关键词:基因组编辑 CRISPR/Cas 工业生物技术 工业生物学
Application of genome editing technology in industrial microorganisms: current status and perspectives
1. Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, China;
2. School of Biological Engineering, Dalian Polytechnic University, Dalian 116034, Liaoning, China
2. School of Biological Engineering, Dalian Polytechnic University, Dalian 116034, Liaoning, China
Abstract: Precise and efficient manipulation of gene expression or rewriting genome sequence is the research hotspots of genome editing, and it is also the core enabling technology contributing to the rapid development of industrial biotechnology. Genome editing technology has experienced three stages of development, from zinc finger nuclease (ZFNs), to transcription activator like effector nuclease (TALEN) and Cas nuclease. Currently, vigorous development of CRISPR/Cas has enabled researchers establish a series of first-generation and second-generation Cas-based genome editing technologies. This contributed to the establishment and optimization for prokaryotic chassis such as Escherichia coli or eukaryotic chassis such as Saccharomyces cerevisiae. This paper summarizes the current development and application of industrial biotechnology using conventional chassis cells, and prospects future development trend with the aim to facilitate researchers to optimize industrial biotechnology and its potential applications.
Keywords:
genome editing CRISPR/Cas industrial biotechnology industrial biology
工业生物学是一门从分子、细胞和系统3个方面解析工业环境下生物体行为的基本规律与作用机制的一门科学,助力工业环境下各类生物体设计及应用等关键科学问题的解决[1-2]。伴随工业生物学概念的提出,工业生物技术的发展也随之而至,在工业生物学这一基础学科的支撑下,工业生物技术迎来蓬勃发展,并为人类经济发展提供所需的基础化学品、医药产品和材料等。目前,工业生物学以分子、细胞和系统3个层面为基础,进一步发展了工业蛋白科学、工业细胞科学及工业发酵科学等方向,研究人员也从蛋白、细胞和发酵3个方向进行优化,以大肠杆菌(Escherichia coli)、酿酒酵母(Saccharomyces cerevisiae) 等工业生物为核心底盘细胞,开发优异的工业化产品。值得注意的是,蛋白、细胞和发酵系统的优化离不开基因组编辑技术的发展,快速且精准的实现工业生物基因组的编辑可助力分子、细胞和系统的迅速进化,加速工业生物技术的发展。
1 基因组编辑技术的发展历程基因组编辑技术是指一类可对生物体基因组进行设计、改造的生物学技术,可实现基因组表达的操控或基因组序列的改写,其发展阶段经历了锌指核酸酶(zinc finger nucleases, ZFNs)、转录激活因子样效应物核酸酶(transcription activator-like effector nucleases, TALEN) 和Cas核酸酶3次技术革新(表 1)。
表 1 ZFN、TALEN和CRISPR/Cas9技术比较
Table 1 Comparison among ZFN, TALEN and CRISPR/Cas9 technologies
| Feature | ZFN | TALEN | CRISPR/Cas9 |
| Efficiency | Low | Low | High |
| Specificity | High | High | High |
| Simplicity | Hard | Hard | Easy |
| Universality | Restrict | Restrict | Wide |
ZFN技术[3-4]主要包括两个组分,一是用于结合特异性DNA序列的含有锌指结构的蛋白质,二是可实现DNA非特异性切割的核酸内切酶Fok Ⅰ,该技术利用一对串联的锌指蛋白靶向DNA序列,从而将内切酶Fok Ⅰ锚定至目标切割位点,进行基因组序列的切割,产生DNA双链断裂。ZFN技术可通过锌指蛋白的数量调整DNA序列结合的特异性,然而靶向的基因组序列和锌指蛋白序列需要复杂的工序进行分析设计,且ZFN相关的技术存在专利封锁,造成该技术至今无法广泛应用。
1.2 转录激活因子样效应物核酸酶技术TALEN技术[5-6]同样由两部分构成,一是来源于转录激活物样效应物的重复序列,可特异性靶向基因组序列,二是介导DNA序列切割的核酸酶Fok Ⅰ。相比于ZFN技术,TALEN技术的设计更为简单,毒性低且效率相当,然而TALEN技术对应的融合蛋白尺寸更大,且重复序列更多,造成其在大肠杆菌等工业生物中组装困难。
1.3 Cas核酸酶CRISPR (clustered regularly interspaced short palindromic repeats)/Cas系统是存在于细菌等微生物体内的一类适应性免疫反应蛋白系统,可用于抵御外源病原体的侵入,CRISPR/Cas系统通过引导RNA (small guide RNA, sgRNA) 靶向识别病原体的基因组,并在Cas核酸酶的作用下破坏病原体的基因组。近年来,随着研究人员对这一系统的认知和改造,CRISPR/Cas系统已被广泛应用于原核生物基因组的编辑,并且由于CRISPR/Cas9技术在基因组编辑及改造中的优越性,研究人员基于该技术优化并进一步发展了多个助力工业生物细胞进化的新型技术,极大促进工业生物技术的发展。
1.3.1 一代CRISPR技术CRISPR/Cas系统包含了靶向基因组序列的sgRNA序列以及行使DNA切割功能的Cas蛋白[7-8],因此,研究人员可直接通过设计成对的sgRNA靶向序列,实现对靶基因序列的双链切割,而切割的DNA双链会通过两种方式进行修复[9],其中非同源末端连接(non-homologous end-joining, NHEJ) 的修复方式会造成目标序列的删除,诱导靶基因的敲除[10-11],而高保真的同源重组修复(homology-directed repair, HDR) 可在外源DNA片段的引入下实现目标序列的插入,诱导靶基因的表达[12-13]。然而,Cas核酸酶诱导的DNA双链断裂会造成基因组的不稳定,且该方法诱导的靶基因敲除和表达效率较低。
1.3.2 二代CRISPR技术研究人员通过对Cas9蛋白分子机制的分析,理性设计了可不诱导DNA双链断裂的dCas9 (dead Cas9) 蛋白,随后将能够发挥转录激活或抑制的效应蛋白与dCas9蛋白进行融合表达,在经由sgRNA的引导下,靶向特定基因的启动子或增强子区域,诱导靶基因的激活或抑制表达。由此,研究人员开发了CRISPRa (CRISPR activation)[14]和CRISPRi (CRISPR interference)[15]技术,可分别诱导基因的转录激活和抑制,CRISPRa和CRISPRi技术虽然特异性强,且效率高,但并不能实现基因组序列的改写。
碱基编辑(base editing, BE) 技术同样利用了核酸酶失活的Cas9突变体(dead Cas9或nickase Cas9),研究人员将Cas9突变体与效应蛋白-脱氨酶蛋白融合表达,构建了碱基编辑系统[16-18]。该技术在脱氨酶和Cas9/sgRNA的作用下可诱导靶基因序列的胞嘧啶、腺嘌呤发生脱氨反应,随后在DNA修复及DNA复制机制的作用下,实现碱基的转换,迄今,碱基编辑系统已经实现了碱基C-T[18]、A-G[17]、C-G[16]、C-A[16]的转换。碱基编辑系统不会诱导DNA双链断裂,也无需外源DNA的引入,即可实现靶基因的突变,且具有设计和操作简便、效率高的特点。
引导编辑(prime editor, PE) 系统包含了发挥逆转录功能的逆转录酶以及核酸酶部分失活的nCas9蛋白,同时在sgRNA的3ʹ端添加了一段引物和模板序列,可实现任意碱基之间的转换及靶基因序列的插入[19-20],但引导编辑的设计较为繁琐,且靶向编辑效率有待进一步的优化。
2 基因组编辑技术在工业生物技术中的应用基因组编辑技术在工业生物技术中的应用载体主要为工业生物底盘细胞,如大肠杆菌、酿酒酵母等微生物,我们将以工业底盘细胞为主,分类介绍基因组编辑技术在工业生物技术的发展历程、应用效果等特点(表 2)。
表 2 工业生物中CRISPR/Cas技术的进展
Table 2 Advances of CRISPR/Cas technologies in industrial microorganisms
| Organism | Type | CRISPR/Cas | Next-generation CRISPR/Cas |
| Escherichia coli | Prokaryote | CRISPR/Cas9, CFPO, CAGO | CBE, DBE, GBE |
| Corynebacterium glutamicum | Prokaryote | CRISP/Cas12, CRISPR/Cas9 | CRISPRi, CBE, MACBETH, ABE, TadA-dCas9-AID, BETTER |
| Bacillus subtilis | Prokaryote | CRISPR/Cas9 | CRISPRi, CBE |
| Streptomyces | Prokaryote | CRISPR/Cas9, CRISP/Cas12 | CRISPRa/i, asRNA-BE |
| Ralstonia eutropha | Prokaryote | CRISPR/Cas9 | |
| Saccharomyces cerevisiae | Eukaryote | CRISPR/Cas9, GTR-CRISPR, mGE | CRISPRa, CBE |
| Myceliophthora | Eukaryote | Cas9/Cas12, CAMR | CBE |
| Aspergillus niger | Eukaryote | CRISPR/Cas9 | CBE |
大肠杆菌是肠杆菌科的一员,现作为细菌的模式生物广泛用于科学研究,不仅如此,随着工业生物技术的发展,大肠杆菌作为工业生物产品的底盘细胞也被广泛应用,如利用大肠杆菌生产β-胡萝卜素[21]、丁二酸[22]等。2013年,洛克菲勒大学Luciano A. Marraffini团队首次应用CRISPR/Cas9系统(Cas9蛋白、双RNA、λ-Red蛋白和单链线性DNA) 对大肠杆菌进行基因组编辑,诱导基因组突变效率可达65%[23];2015年,天津大学赵学明团队对RNA系统简化,并在单个sgRNA的介导下实现大肠杆菌的基因组编辑,最高效率可达100%[21];随后为进一步简化基因组编辑步骤及缩短筛选时间,2017年,中国科学院天津工业生物技术研究所(简称天津工业生物所) 毕昌昊和张学礼团队共同开发了CFPO (CRISPR/Cas9-facilitated multiplex pathway optimization) 技术[24],研究人员首先将pRedCas9质粒和RBS (ribosome binding site) 供体DNA文库共转染至大肠杆菌中,诱导基因重组,随后继续转染gRNA文库质粒,在染色质水平上进一步诱导基因组编辑,提高了优势细胞筛选的效率。CFPO技术使得研究人员可以从染色质水平上同时调控多个基因,与通过质粒同时过表达多个基因相比[25-26],该技术不会给细胞造成生长负担,也可以忽略质粒表达的不稳定性;同年两团队再次合作建立了CAGO (CRISPR/Cas9-assisted gRNA-free one-step) 基因组编辑技术[27],该技术首先借由同源重组的方法将Cas9高效靶向的N20序列提前插入大肠杆菌基因组中,随后通过Cas9蛋白诱导基因组删除、插入的功能,完成高效、特异的基因组编辑。值得注意的是,Cas9蛋白与基因序列特异性的结合受限于前间隔序列邻近基序(protospacer adjacent motif, PAM) 序列的识别[28],而CAGO技术的使用可以忽略Cas9蛋白结合的PAM序列限制,且研究人员通过该技术可高效删除100 kb甚至更大的染色质片段。
为进一步提高CRISPR/Cas9技术在大肠杆菌基因组中的编辑效率,2018年,神户大学Akihiko Kondo团队首次将PmCDA脱氨酶与Cas9蛋白系统融合,建立了大肠杆菌C > T突变的碱基编辑系统,突变效率可达95.1%[29];2019年,天津工业生物所毕昌昊和张学礼团队将脱氨酶TadA和nCas9 (nickase Cas9) 构建的碱基编辑系统与诱导性的活性Cas9融合,开发了DBE (double-check editing) 编辑技术[30],该技术在碱基编辑的基础上同时引入了靶向序列一致的活性Cas9蛋白,可诱导未发生碱基编辑细胞的死亡,进一步提升大肠杆菌中ABE (adenosine base editor) 的编辑效率;2021年,两团队再次合作首次报道了一种新型碱基编辑工具-糖基化酶碱基编辑系统(glycosylase base editor, GBE),可在大肠杆菌中高效实现碱基C > A的突变,极大助力大肠杆菌的基因组编辑[16],该技术将活化诱导的胞苷脱氨酶(activation- induced cytidine deaminase, AID) 与nCas9蛋白融合表达,随后转入大肠杆菌中,实现了大肠杆菌碱基C > A的突变(图 1),效率可达近90%,与既往碱基C > T和A > G突变类型相比[17-18],该技术首次报道了一种新型基因组碱基突变类型C > A突变,丰富了大肠杆菌基因组编辑工具。
谷氨酸棒状杆菌(Corynebacterium glutamicum) 是一类革兰氏阳性菌,是工业生物领域中重要的底盘细胞之一,被广泛用于氨基酸的生物制造[31]。近年来,谷氨酸棒状杆菌基因组编辑技术正在不断发展,不仅推动了谷氨酸棒状杆菌的基础研究,也加速了基于谷氨酸棒状杆菌的细胞工厂的创建和优化。2016年,麻省理工学院Timothy K. Lu团队首次成功应用CRISPR/Cas9系统对谷氨酸棒状杆菌进行基因组编辑,研究人员将失活的dCas9蛋白靶向特定基因组,建立了CRISPRi技术,其抑制单个基因的表达效率可达98%[32];2017年,中国科学院上海生命科学研究院杨晟团队在谷氨酸棒状杆菌中成功将CRISPR/Cas12系统与单链DNA重组系统结合,并介导基因组突变,编辑效率可达86%–100%[33];同年7月,韩国科学技术院Sang Yup Lee团队发现可通过密码子优化的方式降低Cas9蛋白在细菌中的毒性,并首次在谷氨酸棒状杆菌中建立基于CRISPR/Cas9的基因组编辑技术[34];同年11月,天津工业生物所郑平和孙际宾团队在谷氨酸棒状杆菌中也通过重构Cas9和gRNA的表达盒,解决了Cas9毒性和gRNA自身转录终止子无法终止的问题,且研究人员采用双质粒共转化的方法在谷氨酸棒状杆菌中实现了基因敲除和插入(效率可达47.3%)[35],与Cas12介导的基因组编辑相比[33],Cas9诱导基因组富含GC序列的谷氨酸棒状杆菌的编辑范围更为广谱。此外,为提升谷氨酸棒状杆菌基因组的编辑效率和多样性,研究人员还将噬菌体重组酶RecT与CRISPR系统进行联合应用,提升了基因组编辑效率,并进一步优化实现了多基因靶点的高效基因组编辑[33-34, 36]。
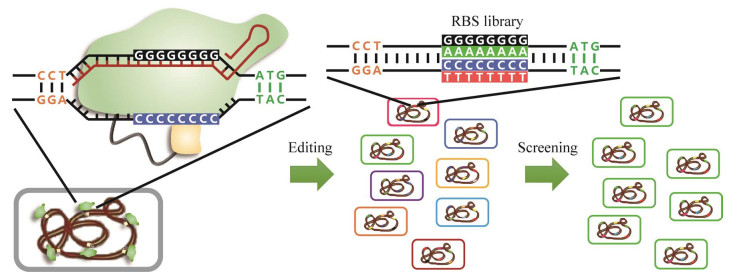
谷氨酸棒状杆菌的同源重组修复效率低下,而碱基编辑技术并不依赖于基因组的同源重组效率,因此,为进一步优化谷氨酸棒状杆菌基因组的编辑类型和编辑效率,2018年,天津工业生物所郑平和孙际宾带领的系统与合成生物技术研究团队与王猛带领的高通量新分子生物合成研究团队合作,在谷氨酸棒状杆菌中开发了基于碱基编辑技术的多元自动化基因组编辑方法(multiplex automated corynebacterium glutamicum base editing method, MACBETH)[37]。研究人员首先结合CRISPR/Cas9系统的定位功能与胞嘧啶脱氨酶(AID) 的碱基编辑功能,可在染色体靶位点特异性实现C > T的编辑,效率高达90%,并由此诱导基因的转录提前终止,从而失活靶基因。随后借助自动化平台,研究团队可实现从质粒构建、基因组编辑、获取正确突变株和表型验证的全流程自动化操作,相比于人工操作的时效性长和不稳定性等缺点,MACBETH技术可实现每月数千突变株的构建,为谷氨酸棒状杆菌的基因组改造提供强大的技术支持。然而,该技术仍存在靶标范围、编辑窗口和碱基转化种类有限的问题,且人工设计碱基编辑所需sgRNA的过程繁琐,限制了全流程自动化的碱基编辑。2019年研究团队再次合作,并对谷氨酸棒状杆菌的碱基编辑工具进行了扩展[38]。首先,研究人员利用识别不同PAM的Cas突变体,将PAM限制由NGG扩展为NG,使可编辑的靶点数量提高了3.9倍;第二,通过截短或延长gRNA长度,将编辑窗口由原来的5 bp扩展为7 bp;第三,在CRISPR/Cas系统上引入腺苷脱氨酶,建立了A到G的腺嘌呤碱基编辑工具;最后,研究人员为精简sgRNA的设计流程,开发了在线工具,方便了碱基编辑工具在基因失活应用中sgRNA的设计与评估,该研究进一步丰富了谷氨酸棒状杆菌基因组编辑工具库;2020年,江南大学刘龙团队将胞嘧啶脱氨酶和腺嘌呤脱氨酶进行融合,在谷氨酸棒状杆菌中开发了双碱基编辑系统,可同时实现C > T/G和A > G的基因组编辑[39];不仅如此,研究人员还将碱基编辑系统应用于基因组的转录调控研究,2022年,郑平等团队再次合作开发了基于碱基编辑的多基因精细表达调控新技术BETTER (base editor-targeted and template-free expression regulation)[40],创新性地将碱基编辑用于基因组原位表达调控元件建库,在谷氨酸棒状杆菌中首次实现了10个基因的组合表达调控,BETTER技术的设计理念是预先在染色体中目的基因上游引入特定的转录或翻译元件(如富含G的RBS序列),应用碱基编辑器编辑产生转录或翻译元件多样性,后根据目的表型进行筛选富集,以获得符合调控目的的突变菌株(图 2)。该技术颠覆了传统的基因表达调控方法[24],突破了组合调控对整合模板的依赖,降低了对外源片段转化效率的要求,可在一轮实验中完成多基因表达调控,有望在多种难编辑工业细胞体内实现复杂的代谢调控,为复杂的代谢调控模式设计开拓新方向。
枯草芽孢杆菌(Bacillus subtilis) 是一种被大量研究的革兰氏阳性菌,被广泛应用于抗生素、药用蛋白、工业酶和生物聚合物的生产,因其优良的蛋白质分泌能力,常被用作工业生产中的重要底盘细胞之一[41]。2016年,滑铁卢大学C. Perry Chou团队首次在枯草芽孢杆菌中建立了CRISPR/Cas9系统,包括基于活性Cas9的基因突变和基于dCas9的CRISPRi基因转录调控工具,并成果实现单基因和双基因的基因组编辑,效率可达100%和85%[42];2019年,为进一步提升基因组编辑效率及减少活性Cas9蛋白双链切割的副反应,天津大学赵学明团队开发了基于nCas9蛋白的基因组编辑技术,该技术的整体编辑效率高于活性Cas9技术,尤其在多基因的编辑中效果提升更为明显[43]。
2020年,天津工业生物所王猛和毕昌昊团队与爱丁堡大学Susan J. Rosser实验室合作,利用dCas9 (dead Cas9) 和AID脱氨酶在枯草芽孢杆菌中首次开发了枯草芽孢杆菌的基因组碱基编辑方法,实现了基因组碱基C > T的编辑,并在多基因组编辑中实现高效应用[44]。该碱基编辑系统在枯草芽孢杆菌168菌株中有5 nt的编辑框,位于PAM上游–16至–20区域,其中–18位点的编辑效率最好,最高能够达到100%。此外,该方法还能同时实现3–4个基因的同时高效编辑,为枯草芽孢杆菌的基因操作和工业应用提供有效工具。
2.4 链霉菌基因组编辑技术进展链霉菌(Streptomyces) 是许多重要天然产物的生产者,其基因组蕴含着大量未被开发的次级代谢生物合成基因簇,也是工业生物重要的底盘细胞之一[45]。2015年,伊利诺伊大学赵惠民团队在链霉菌中首次报道了基于CRISRP/Cas9的基因组编辑技术,并在3种类型的链霉菌中实现染色质片段的删除,编辑效率在70%–100%[46];2020年,天津大学罗云孜团队在链霉菌中开发了基于CRISPR/Cas12a的基因组编辑技术,该技术不仅能对长达100 kb的基因组片段进行高效删除,同时研究人员发现了一个PAM框更为广谱的Cas突变体,进一步优化了编辑工具的应用范围[47]。
2022年,为提升基因组中低表达生物合成基因簇的表达效率,莱斯大学James Chappell团队在链霉菌中将dCas9蛋白与转录激活结构域融合,开发了CRISPRa技术,并通过靶向基因组转录起始及延伸的sgRNA优化了现有的CRISPRi技术,研究人员还将这两种技术用于链霉菌内源性调控网络的基因表达调控,成功实现沉默生物合成基因簇的高效表达[48]。
基于CRISPR/Cas9发展起来的单碱基编辑技术也已成功应用于天蓝色链霉菌等一些模式菌株中,相较于传统CRISPR技术更为方便快捷。基于胞嘧啶脱氨酶的碱基编辑效率与底盘细胞尿嘧啶DNA糖苷酶(uracil DNA glycosylase, UDG) 的功能密切相关,枯草芽孢杆菌研究人员前期多用噬菌体来源的尿嘧啶DNA糖苷酶抑制剂(uracil glycosylase inhibitor, UGI) 来提升碱基编辑效率,但在某些情况下,其抑制效果却并不理想。2021年,天津工业生物所王猛研究员带领的高通量编辑与筛选平台实验室,与天津科技大学花尔并教授团队合作,在模式菌株变铅青链霉菌(Streptomyces lividans) 66中研究了UDG与碱基编辑效率的关系,并开发了新一代碱基编辑器asRNA-BE (antisense RNA interference-enhanced CRISPR/Cas9 Base editing method)[49]。研究人员首先利用融合了胞苷脱氨酶rAPOBEC1的BE系列碱基编辑器成功实现基因组3个次级代谢基因的编辑。在此基础上,研究团队利用CRISPR/Cas9辅助的基因敲除手段,分别构建了尿嘧啶DNA糖苷酶UDG1和UDG2的单敲和双敲菌株,发现在失活UDG的情况下,碱基编辑效率可较原始提高3.4倍到67.4倍不等。考虑到UDG是细胞修复过程中的关键酶,其长期缺失不利于菌株基因组的稳定,团队利用反义RNA干扰降低基因表达水平的策略代替基因敲除,在原始的BE编辑器基础上整合了针对UDG的反义RNA干扰模块,构建了新一代碱基编辑器asRNA-BE,成功将编辑效率提高至原来的2.8倍到65.8倍不等。asRNA-BE编辑器可以在碱基编辑过程中瞬时抑制UDG的表达。基因组测序结果也表明,与BE编辑器相比,asRNA-BE编辑器在大幅度提高编辑效率的同时,并未引起额外的脱靶,在工业细胞改造等方面具有良好的应用前景。
2.5 罗氏菌基因组编辑技术进展罗氏菌(Ralstonia eutropha) 是一种广泛存在于土壤和淡水环境中的革兰氏阴性杆菌,近年来,随着研究人员对其经济价值的挖掘,罗氏菌也被开始用于设计生产如支链醇等生物燃料[50]。目前,罗氏菌基因组编辑技术主要依赖于Ⅱ型内含子和自杀性质粒介导的编辑,但这2种方法设计复杂、耗时且效率低下[51]。2018年,中科院工业生物所毕昌昊和张学礼研究团队首次在罗氏菌中应用了CRISPR/Cas9基因组编辑技术,成功开发了基于Cas9编辑的基因组插入技术[52],为将Cas9基因组编辑技术应用于罗氏菌,研究人员首先对罗氏菌的电穿孔效率进行优化提高质粒转化效率,随后利用阿拉伯糖诱导型启动子表达Cas9蛋白介导基因组编辑,最后研究人员在罗氏菌中共对5个基因进行了分别编辑,效率可达78.3%–100%。
2.6 酿酒酵母基因组编辑技术进展酿酒酵母作为工业生物领域中重要的底盘细胞之一,被广泛用于开发传统酿造食品、大宗化学品和高附加值工业产品[53],而基因组编辑技术的发展同样为加快酿酒酵母细胞相关工业产品的开发提供强有力的使能技术。早在2013年,哈佛医学院George M. Church团队便在酿酒酵母中建立了CRISPR/Cas9基因组编辑工具,研究人员发现在供体DNA添加的情况下,Cas9和瞬时表达的gRNA盒介导的DNA双链断裂最高可提升细胞同源重组效率达130倍,而在持续过表达Cas9和gRNA表达质粒后,同源重组编辑效率可达100%[54];虽然多个基因的连续性编辑可以依赖选择性标记物的筛选,但标记物的回收耗时耗力,为进一步提升多基因同时编辑的效率,研究人员对gRNA表达系统进行优化,以实现酵母基因组多基因的高效编辑。2015年,丹麦技术大学Jay D.Keasling团队使用单个SNR52启动子在同一质粒中启动多个gRNA的表达,虽然编辑效率较高,但转化效率出现明显下降[55];2019年,北京化工大学刘子鹤团队建立了GTR-CRISPR (gRNA-tRNA array for CRISPR-Cas9) 系统,研究人员在不同的gRNA表达序列之间加入了tRNAGly序列,从而实现多个gRNA序列的同时等效表达,并在该技术的支持下,最高实现8个基因的同时编辑,效率可达87%[56];2022年,考虑到酿酒酵母NHEJ修复途径介导的基因组突变能力较差,天津工业生物所王钦宏团队基于对酿酒酵母内源双链DNA断裂修复途径的改造,将筛选的DNA修复蛋白与Cas9蛋白融合,建立了不依赖模板的高效致突变基因组技术(mutagenic genome editing, mGE)[57],与CRISPR/Cas9常规诱导的基因组突变[58-59]相比,该技术显著提升了酿酒酵母基因组突变的多样性,实现了靶向基因的可遗传多样性调控表达。研究人员通过评价和比较DNA修复途径中多个关键基因的敲除、过表达或氨基酸突变等对CRISPR/Cas系统诱导基因组编辑功能的影响,构建了基于MRE11-H125N (MRE11核酸酶活性失活的等位基因) 和Cas9/Cpf1的编辑工具组合,并在酿酒酵母中通过调节FPS1和GPD1的表达有效提高了乙醇生产能力。
2017年,德克萨斯大学Hal S Alper团队在酿酒酵母中应用了CRISPRa技术实现了基因组的阶级表达水平,研究人员采用了dCas9-VPR编辑器,并使用靶向目标基因启动子不同位置的gRNA序列,结果发现,dCas9-VPR介导的基因组表达水平与gRNA靶向序列和启动子的距离正相关,由此研究人员进一步利用该系统鉴定了酿酒酵母中关键代谢途径的限速步骤,从而提升甘油生产能力[60]。
2022年,天津工业生物所毕昌昊和张学礼研究团队解析了酿酒酵母中碱基C > G突变的分子机制,并以此为基础开发了基于不同DNA聚合酶的碱基编辑系统,该系统在依赖于不同聚合酶的调控下,可在酵母中分别特异性实现碱基C > G和C > A的编辑[61],为酵母基因组编辑提供高效、便捷的工具。
2.7 嗜热毁丝霉基因组编辑技术进展嗜热毁丝霉(Myceliophthora) 是一种能够快速降解纤维素的嗜热丝状真菌,其分泌的纤维素水解酶的种类和数量丰富,且稳定性好,因此该菌在纤维素酶生产和生物基燃料的研发方面具有巨大潜力[62]。2017年,天津工业生物所田朝光团队以嗜热毁丝霉为研究对象,采用自主挖掘的嗜热毁丝霉U6启动子,诱导sgRNA的转录表达,并采用原生质体共转化的方法将编辑系统和同源重组模板导入到目标细胞中,首次建立了CRISPR/Cas9介导的嗜热真菌基因组编辑系统[63],与既往通过RNA干扰实现的基因表达敲低效率(约20%)[64]相比,CRISPR介导的单基因缺失的同源重组效率高达90%–95%。Cas12蛋白是一类CRISPR系统中的Ⅴ型核酸酶,因其诱导基因突变的能力同样被广泛应用于各类工业生物,虽然Cas12核酸酶的应用已在谷氨酸棒状杆菌[33]、链霉菌[65]等微生物中报道,但Cas12在嗜热真菌中的应用还未有报道。2019年,该团队研究人员进一步升级了嗜热真菌基因组编辑技术,构建了一种基于Ⅴ型Cas12a核酸酶的新型基因组编辑体系[66],并将其与crRNA-array串联表达系统联合,从而能够快速、简易地实现基因组多基因的编辑,此外,研究人员还将Cas12a系统和Cas9系统的交替使用,进一步提高靶点选择的灵活性。不仅如此,该团队基于嗜热真菌CRISPR-Cas9/Cas12a编辑系统,交替使用2个选择性标记基因neo和bar,进一步构建了标记基因回收和交替使用的丝状真菌CRISPR-Cas基因组编辑技术系统(CRISPR-Cas-assisted marker recycling, CAMR)[66],丰富了工业丝状真菌基因组编辑系统选择的多样性。
2022年,田朝光团队通过对APOBEC1、CDA脱氨酶的进化,在嗜热毁丝霉中成功开发了胞嘧啶单碱基编辑系统,可有效实现碱基C > T的置换,随后研究人员还通过碱基编辑诱导终止密码子的功能成功实现amdS基因和cre-1基因的失活,编辑效率最高可达92%[67],该技术为快速、高效且精准的创建嗜热真菌突变体提供有力工具。
2.8 黑曲霉基因组编辑技术进展黑曲霉(Aspergillus niger) 是重要的传统工业生物细胞,在有机酸与酶制剂等领域直接产值近百亿美元。黑曲霉具有极端环境耐受性、高生产经济性、强发酵鲁棒性和食品安全性等特性,是不可多得的理想细胞工厂[68]。2017年,自然资源和生命科学大学的Matthias G. Steiger团队在黑曲霉中报道了基于CRISPR/Cas9的基因组编辑技术,研究人员使用了RNA聚合酶Ⅱ启动子诱导sgRNA的表达,基因组整合的编辑效率最高可达100%[69];2018年,天津工业生物所孙际宾与郑平研究团队通过对异源性及黑曲霉内源性U6启动子的挖掘,首次报道了基于U6启动子表达sgRNA的CRISPR/ Cas9技术[70],与既往通过RNA聚合酶Ⅱ启动子诱导的sgRNA表达系统相比[69],该技术在整体应用设计上更为灵活,进一步丰富黑曲霉基因组编辑技术进展;2019年,两团队为解决CRISPR/Cas9系统中缺乏普适性的高效表达sgRNA这一问题,首次提出以5S rRNA的启动子来驱动sgRNA表达的策略[71],研究发现5S rRNA启动子可显著提高黑曲霉中sgRNA的表达丰度,引导Cas9蛋白靶向特定基因位点进行切割,从而使基于NHEJ系统的基因失活效率达100%。不仅如此,研究人员发现在NHEJ失活的底盘细胞中利用“two-cuts”的基因精准敲除策略进行一次基因组编辑,即可实现长至48 kb的基因簇敲除,极大提升了黑曲霉基因组编辑效率。
2019年,华南理工大学潘力团队首次报道了一种在黑曲霉中建立的碱基编辑系统[72],可高效诱导基因组C > T的碱基编辑,实验中研究人员使用了rAPOBEC1和nCas9蛋白建立了碱基编辑系统,编辑窗口达8个碱基,此外,研究人员还通过该技术诱导了尿苷营养缺陷型基因和色素基因的失活,编辑效率可达47.36%–100%,该技术进一步丰富了黑曲霉基因组编辑工具文库。
3 基因组编辑技术的展望伴随CRISPR相关基因组编辑技术的不断发展,可以预见工业生物技术的发展也会迎来爆发期,然而目前基于CRISPR发展的基因组编辑技术在工业生物领域中的应用潜力还有待进一步挖掘。首先,基于CRISPR开发的基因组编辑技术多在真核生物领域中加以应用,在微生物相关的工业生物领域中的应用还少有研究,如先导编辑技术、RNA编辑技术和转座酶编辑技术等,目前在酿酒酵母、丝状真菌等工业生物基因组编辑中所需的逆转录酶、RNA编辑催化酶等仍需进一步挖掘,且这些技术助力基因组编辑的效率、方式等也需研究工作探索;其次,伴随工业生物细胞队伍的不断壮大,对于新型工业基因组编辑技术的开发及发展也是工业生物技术研究中的重点,此外对于现阶段基因组编辑效率、特异性较低的技术也要进一步地优化革新,从而保证工业生物技术的多样性发展;最后,随着生物信息技术的不断进步和人工智能技术的快速发展,新型基因组编辑技术的开发以及基于人工智能这一新模式开发的基因组编辑技术也值得期待。
参考文献
| [1] |
Straathof AJJ, Wahl SA, Benjamin KR, et al. Grand research challenges for sustainable industrial biotechnology. Trends Biotechnol, 2019, 37(10): 1042-1050. DOI:10.1016/j.tibtech.2019.04.002
|
| [2] |
Clomburg JM, Crumbley AM, Gonzalez R. Industrial biomanufacturing: the future of chemical production. Science, 2017, 355(6320): aag0804. DOI:10.1126/science.aag0804
|
| [3] |
Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok Ⅰ cleavage domain. PNAS, 1996, 93(3): 1156-1160. DOI:10.1073/pnas.93.3.1156
|
| [4] |
Smith J, Bibikova M, Whitby FG, et al. Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res, 2000, 28(17): 3361-3369. DOI:10.1093/nar/28.17.3361
|
| [5] |
Boch J, Scholze H, Schornack S, et al. Breaking the code of DNA binding specificity of TAL-type Ⅲ effectors. Science, 2009, 326(5959): 1509-1512. DOI:10.1126/science.1178811
|
| [6] |
Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science, 2009, 326(5959): 1501. DOI:10.1126/science.1178817
|
| [7] |
Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science, 2012, 337(6096): 816-821. DOI:10.1126/science.1225829
|
| [8] |
Barrangou R. RNA-mediated programmable DNA cleavage. Nat Biotechnol, 2012, 30(9): 836-838. DOI:10.1038/nbt.2357
|
| [9] |
Paquet D, Kwart D, Chen A, et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature, 2016, 533(7601): 125-129. DOI:10.1038/nature17664
|
| [10] |
Sakuma T, Nakade S, Sakane Y, et al. MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat Protoc, 2016, 11(1): 118-133. DOI:10.1038/nprot.2015.140
|
| [11] |
Mandl M, Ritthammer H, Ejaz A, et al. CRISPR/Cas9-mediated gene knockout in human adipose stem/progenitor cells. Adipocyte, 2020, 9(1): 626-635. DOI:10.1080/21623945.2020.1834230
|
| [12] |
Richardson CD, Ray GJ, DeWitt MA, et al. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol, 2016, 34(3): 339-344. DOI:10.1038/nbt.3481
|
| [13] |
Albadri S, del Bene F, Revenu C. Genome editing using CRISPR/Cas9-based knock-in approaches in zebrafish. Methods, 2017, 121/122: 77-85. DOI:10.1016/j.ymeth.2017.03.005
|
| [14] |
Konermann S, Brigham MD, Trevino AE, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature, 2015, 517(7536): 583-588. DOI:10.1038/nature14136
|
| [15] |
Gilbert LA, Larson MH, Morsut L, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell, 2013, 154(2): 442-451. DOI:10.1016/j.cell.2013.06.044
|
| [16] |
Zhao D, Li J, Li S, et al. Glycosylase base editors enable C-to-A and C-to-G base changes. Nat Biotechnol, 2021, 39(1): 35-40. DOI:10.1038/s41587-020-0592-2
|
| [17] |
Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A·T to G·C in genomic DNA without DNA cleavage. Nature, 2017, 551(7681): 464-471. DOI:10.1038/nature24644
|
| [18] |
Komor AC, Kim YB, Packer MS, et al. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature, 2016, 533(7603): 420-424. DOI:10.1038/nature17946
|
| [19] |
Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature, 2019, 576(7785): 149-157. DOI:10.1038/s41586-019-1711-4
|
| [20] |
Chen PJ, Hussmann JA, Yan J, et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell, 2021, 184(22): 5635-5652. DOI:10.1016/j.cell.2021.09.018
|
| [21] |
Li Y, Lin Z, Huang C, et al. Metabolic engineering of Escherichia coli using CRISPR-Cas9 meditated genome editing. Metab Eng, 2015, 31: 13-21. DOI:10.1016/j.ymben.2015.06.006
|
| [22] |
Zhang W, Zhang T, Song M, et al. Metabolic engineering of Escherichia coli for high yield production of succinic acid driven by methanol. ACS Synth Biol, 2018, 7(12): 2803-2811. DOI:10.1021/acssynbio.8b00109
|
| [23] |
Jiang WY, Bikard D, Cox D, et al. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol, 2013, 31(3): 233-239. DOI:10.1038/nbt.2508
|
| [24] |
Zhu X, Zhao D, Qiu H, et al. The CRISPR/Cas9-facilitated multiplex pathway optimization (CFPO) technique and its application to improve the Escherichia coli xylose utilization pathway. Metab Eng, 2017, 43(pt a): 37-45.
|
| [25] |
Yin LH, Zhao JX, Chen C, et al. Enhancing the carbon flux and NADPH supply to increase L-isoleucine production in Corynebacterium glutamicum. Biotechnol Bioprocess Eng, 2014, 19(1): 132-142. DOI:10.1007/s12257-013-0416-z
|
| [26] |
Zelcbuch L, Antonovsky N, Bar-Even A, et al. Spanning high-dimensional expression space using ribosome-binding site combinatorics. Nucleic Acids Res, 2013, 41(9): e98. DOI:10.1093/nar/gkt151
|
| [27] |
Zhao DD, Feng X, Zhu XN, et al. CRISPR/Cas9-assisted gRNA-free one-step genome editing with no sequence limitations and improved targeting efficiency. Sci Rep, 2017, 7: 16624. DOI:10.1038/s41598-017-16998-8
|
| [28] |
Zhang H, Cheng QX, Liu AM, et al. A novel and efficient method for bacteria genome editing employing both CRISPR/Cas9 and an antibiotic resistance cassette. Front Microbiol, 2017, 8: 812. DOI:10.3389/fmicb.2017.00812
|
| [29] |
Banno S, Nishida K, Arazoe T, et al. Deaminase-mediated multiplex genome editing in Escherichia coli. Nat Microbiol, 2018, 3(4): 423-429. DOI:10.1038/s41564-017-0102-6
|
| [30] |
Xin X, Li J, Zhao D, et al. Double-check base editing for efficient A to G conversions. ACS Synth Biol, 2019, 8(12): 2629-2634. DOI:10.1021/acssynbio.9b00284
|
| [31] |
Tsuge Y, Matsuzawa H. Recent progress in production of amino acid-derived chemicals using Corynebacterium glutamicum. World J Microbiol Biotechnol, 2021, 37(3): 49. DOI:10.1007/s11274-021-03007-4
|
| [32] |
Cleto S, Jensen JV, Wendisch VF, et al. Corynebacterium glutamicum metabolic engineering with CRISPR interference (CRISPRi). ACS Synth Biol, 2016, 5(5): 375-385. DOI:10.1021/acssynbio.5b00216
|
| [33] |
Jiang Y, Qian FH, Yang JJ, et al. CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat Commun, 2017, 8: 15179. DOI:10.1038/ncomms15179
|
| [34] |
Cho JS, Choi KR, Prabowo CPS, et al. CRISPR/Cas9-coupled recombineering for metabolic engineering of Corynebacterium glutamicum. Metab Eng, 2017, 42: 157-167. DOI:10.1016/j.ymben.2017.06.010
|
| [35] |
Liu J, Wang Y, Lu Y, et al. Development of a CRISPR/Cas9 genome editing toolbox for Corynebacterium glutamicum. Microb Cell Fact, 2017, 16(1): 205. DOI:10.1186/s12934-017-0815-5
|
| [36] |
Liu J, Wang Y, Zheng P, et al. CRISPR/Cas9-mediated ssDNA recombineering in Corynebacterium glutamicum. Bio Protoc, 2018, 8(19): e3038.
|
| [37] |
Wang Y, Liu Y, Liu J, et al. MACBETH: multiplex automated Corynebacterium glutamicum base editing method. Metab Eng, 2018, 47: 200-210. DOI:10.1016/j.ymben.2018.02.016
|
| [38] |
Wang Y, Liu Y, Li JW, et al. Expanding targeting scope, editing window, and base transition capability of base editing in Corynebacterium glutamicum. Biotechnol Bioeng, 2019, 116(11): 3016-3029. DOI:10.1002/bit.27121
|
| [39] |
Deng C, Lv X, Li J, et al. Development of a DNA double-strand break-free base editing tool in Corynebacterium glutamicum for genome editing and metabolic engineering. Metab Eng Commun, 2020, 11: e00135. DOI:10.1016/j.mec.2020.e00135
|
| [40] |
Wang Y, Cheng HJ, Liu Y, et al. In-situ generation of large numbers of genetic combinations for metabolic reprogramming via CRISPR-guided base editing. Nat Commun, 2021, 12: 678. DOI:10.1038/s41467-021-21003-y
|
| [41] |
Liu Y, Liu L, Li J, et al. Synthetic biology toolbox and chassis development in Bacillus subtilis. Trends Biotechnol, 2019, 37(5): 548-562. DOI:10.1016/j.tibtech.2018.10.005
|
| [42] |
Westbrook AW, Moo-Young M, Chou CP. Development of a CRISPR-Cas9 tool kit for comprehensive engineering of Bacillus subtilis. Appl Environ Microbiol, 2016, 82(16): 4876-4895. DOI:10.1128/AEM.01159-16
|
| [43] |
Liu D, Huang C, Guo J, et al. Development and characterization of a CRISPR/Cas9n-based multiplex genome editing system for Bacillus subtilis. Biotechnol Biofuels, 2019, 12: 197. DOI:10.1186/s13068-019-1537-1
|
| [44] |
Yu S, Price MA, Wang Y, et al. CRISPR-dCas9 mediated cytosine deaminase base editing in Bacillus subtilis. ACS Synth Biol, 2020, 9(7): 1781-1789. DOI:10.1021/acssynbio.0c00151
|
| [45] |
Lee N, Hwang S, Lee Y, et al. Synthetic biology tools for novel secondary metabolite discovery in Streptomyces. J Microbiol Biotechnol, 2019, 29(5): 667-686. DOI:10.4014/jmb.1904.04015
|
| [46] |
Cobb RE, Wang YJ, Zhao HM. High-efficiency multiplex genome editing of Streptomyces species using an engineered CRISPR/Cas system. ACS Synth Biol, 2015, 4(6): 723-728. DOI:10.1021/sb500351f
|
| [47] |
Zhang J, Zhang D, Zhu J, et al. Efficient multiplex genome editing in Streptomyces via engineered CRISPR-Cas12a systems. Front Bioeng Biotechnol, 2020, 8: 726. DOI:10.3389/fbioe.2020.00726
|
| [48] |
Ameruoso A, Villegas Kcam MC, Cohen KP, et al. Activating natural product synthesis using CRISPR interference and activation systems in Streptomyces. Nucleic Acids Res, 2022, 50(13): 7751-7760. DOI:10.1093/nar/gkac556
|
| [49] |
Zhang Y, Yun KY, Huang HM, et al. Antisense RNA interference-enhanced CRISPR/Cas9 base editing method for improving base editing efficiency in Streptomyces lividans 66. ACS Synth Biol, 2021, 10(5): 1053-1063. DOI:10.1021/acssynbio.0c00563
|
| [50] |
Li H, Opgenorth PH, Wernick DG, et al. Integrated electromicrobial conversion of CO2 to higher alcohols. Science, 2012, 335(6076): 1596. DOI:10.1126/science.1217643
|
| [51] |
Park JM, Jang YS, Kim TY, et al. Development of a gene knockout system for Ralstonia eutropha H16 based on the broad-host-range vector expressing a mobile group Ⅱ intron. FEMS Microbiol Lett, 2010, 309(2): 193-200.
|
| [52] |
Oh EJ, Jin YS. Engineering of Saccharomyces cerevisiae for efficient fermentation of cellulose. FEMS Yeast Res, 2020, 20(1): foz089. DOI:10.1093/femsyr/foz089
|
| [53] |
DiCarlo JE, Norville JE, Mali P, et al. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res, 2013, 41(7): 4336-4343. DOI:10.1093/nar/gkt135
|
| [54] |
Jakočiūnas T, Bonde I, Herrgård M, et al. Multiplex metabolic pathway engineering using CRISPR/Cas9 in Saccharomyces cerevisiae. Metab Eng, 2015, 28: 213-222. DOI:10.1016/j.ymben.2015.01.008
|
| [55] |
Zhang YP, Wang J, Wang ZB, et al. A gRNA-tRNA array for CRISPR-Cas9 based rapid multiplexed genome editing in Saccharomyces cerevisiae. Nat Commun, 2019, 10: 1053. DOI:10.1038/s41467-019-09005-3
|
| [56] |
Wang Z, Lin Y, Dai Z, et al. Modulating DNA repair pathways to diversify genomic alterations in Saccharomyces cerevisiae. Microbiol Spectr, 2022, 10(2): e0232621. DOI:10.1128/spectrum.02326-21
|
| [57] |
Bao Z, Xiao H, Liang J, et al. Homology-integrated CRISPR-Cas (HI-CRISPR) system for one-step multigene disruption in Saccharomyces cerevisiae. ACS Synth Biol, 2015, 4(5): 585-594. DOI:10.1021/sb500255k
|
| [58] |
Pawelczak KS, Gavande NS, VanderVere-Carozza PS, et al. Modulating DNA repair pathways to improve precision genome engineering. ACS Chem Biol, 2018, 13(2): 389-396. DOI:10.1021/acschembio.7b00777
|
| [59] |
Deaner M, Alper HS. Systematic testing of enzyme perturbation sensitivities via graded dCas9 modulation in Saccharomyces cerevisiae. Metab Eng, 2017, 40: 14-22. DOI:10.1016/j.ymben.2017.01.012
|
| [60] |
Jiang G, Wang J, Zhao D, et al. Molecular mechanism of the cytosine CRISPR base editing process and the roles of translesion DNA polymerases. ACS Synth Biol, 2021, 10(12): 3353-3358. DOI:10.1021/acssynbio.1c00293
|
| [61] |
Wösten HAB. Filamentous fungi for the production of enzymes, chemicals and materials. Curr Opin Biotechnol, 2019, 59: 65-70. DOI:10.1016/j.copbio.2019.02.010
|
| [62] |
Liu Q, Gao R, Li J, et al. Development of a genome-editing CRISPR/Cas9 system in thermophilic fungal Myceliophthora species and its application to hyper-cellulase production strain engineering. Biotechnol Biofuels, 2017, 10: 1. DOI:10.1186/s13068-016-0693-9
|
| [63] |
Yang F, Gong Y, Liu G, et al. Enhancing cellulase production in thermophilic fungus Myceliophthora thermophila ATCC 42464 by RNA interference of cre1 gene expression. J Microbiol Biotechnol, 2015, 25(7): 1101-1107. DOI:10.4014/jmb.1501.01049
|
| [64] |
Li L, Wei K, Zheng G, et al. CRISPR-Cpf1-assisted multiplex genome editing and transcriptional repression in Streptomyces. Appl Environ Microbiol, 2018, 84(18): e00827-18.
|
| [65] |
Liu Q, Zhang Y, Li F, et al. Upgrading of efficient and scalable CRISPR-Cas-mediated technology for genetic engineering in thermophilic fungus Myceliophthora thermophila. Biotechnol Biofuels, 2019, 12: 293. DOI:10.1186/s13068-019-1637-y
|
| [66] |
Zhang C, Li N, Rao L, et al. Development of an efficient C-to-T base-editing system and its application to cellulase transcription factor precise engineering in thermophilic fungus Myceliophthora thermophila. Microbiol Spectr, 2022, 10(3): e0232121. DOI:10.1128/spectrum.02321-21
|
| [67] |
Park HS, Jun SC, Han KH, et al. Diversity, application, and synthetic biology of industrially important Aspergillus fungi. Adv Appl Microbiol, 2017, 100: 161-202.
|
| [68] |
Sarkari P, Marx H, Blumhoff ML, et al. An efficient tool for metabolic pathway construction and gene integration for Aspergillus niger. Bioresour Technol, 2017, 245(pt b): 1327-1333.
|
| [69] |
Zheng X, Zheng P, Sun J, et al. Heterologous and endogenous U6 snRNA promoters enable CRISPR/Cas9 mediated genome editing in Aspergillus niger. Fungal Biol Biotechnol, 2018, 5: 2. DOI:10.1186/s40694-018-0047-4
|
| [70] |
Zheng X, Zheng P, Zhang K, et al. 5S rRNA promoter for guide RNA expression enabled highly efficient CRISPR/Cas9 genome editing in Aspergillus niger. ACS Synth Biol, 2019, 8(7): 1568-1574. DOI:10.1021/acssynbio.7b00456
|
| [71] |
Huang L, Dong H, Zheng J, et al. Highly efficient single base editing in Aspergillus niger with CRISPR/Cas9 cytidine deaminase fusion. Microbiol Res, 2019, 223/224/225: 44-50.
|