
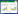


手性医药化学品的绿色生物合成
陈曦
,
吴凤礼
,
樊飞宇
,
吴洽庆
,
朱敦明
生物工程学报  2022, Vol. 38 Issue (11): 4240-4262 2022, Vol. 38 Issue (11): 4240-4262 |







引用本文

|

医药化学品的研发和制造水平关系到人类健康、社会稳定和环境保护等诸多社会问题,同时也是衡量一个国家社会和科技发展水平的重要标准之一。手性医药化学品是已开发的医药化学品中的重要组成部分,根据各制药公司2021年财报披露的产品销售数据,2021年全球药品销售额TOP200的小分子药品中,手性小分子药物占比达60%以上(https://njardarson.lab.arizona.edu/content/top-pharmaceuticals-poster)。目前,手性医药化学品的化学催化往往是其底物合成较困难,放大到工业化中手性催化剂成本较高,且催化剂在反应过程中容易中毒失效,会造成产品光学纯度降低等问题。
生物催化作为新一代工业生物技术的关键技术,具有反应条件温和、催化效率高、选择性好、副产物少、能耗低、催化剂无毒且可降解等特征,受到学术界和工业领域的高度关注,同时也成为各国科技与产业发展的战略重点。近年来,报道了许多生物催化技术应用于合成手性医药化合物的成功案例,极大地推动了医药化学品的绿色合成[1-5]。本综述将基于手性医药化学品的结构特点,按照手性胺、手性醇、天然/非天然氨基酸、甾体化合物、芳香类化合物分类,重点介绍中国科学院天津工业生物技术研究所在此方面的研究进展。
1 手性胺类医药中间体的绿色生物合成在全球药品销售额TOP200的小分子药品中有40个药物分子具有手性胺结构(不包含手性α、β和γ-氨基酸,此内容在3.1–3.4介绍)。手性胺不仅在药物合成中至关重要,其还可作为手性配体使用,因此手性胺的合成一直是不对称催化合成的热点,同时也是生物催化的重要研究内容之一。本节将介绍含有手性胺模块的药物中间体的生物合成进展。
1.1 镇咳药右美沙芬关键手性胺中间体的合成氢溴酸右美沙芬已广泛用作非阿片类镇咳药超过50年,具有抗惊厥和神经保护的特性且不会成瘾,适用于感冒、急性或慢性支气管炎、支气管哮喘、咽喉炎、肺结核以及其他上呼吸道感染引起的干咳,是许多感冒药物中的有效成分[6-7]。右美沙芬(dextromethorphan) 合成中的关键手性中间体为(S)-1-(4-甲氧基苄基)-1, 2, 3, 4, 5, 6, 7, 8-八氢异喹啉((S)-1, 图 1A)[8-9]。工业生产光学纯(S)-1主要是化学拆分法[10-11],但最大理论产率仅有50%,在经济及生态上不理想;过渡金属不对称氢化法则存在产物光学纯度不足(86% ee)、反应条件剧烈及重金属残留等问题[12-13]。
|
|
| 图 1 (S)-1作为手性中间体合成右美沙芬及酶-化学法、酶法合成(S)-1 Fig. 1 (S)-1 as intermediate for the synthesis of dextromethorphan, and chemo-enzymatic and enzymatic synthesis of (S)-1. (A) Synthesis of dextromethorphan using (S)-1 as intermediate. (B) Synthesis of dextromethorphan intermediate (S)-1 catalyzed by cyclohexylamine oxidase CHAO Y321I and NH3·BH3 or imine reductase IR40[14-15]. A:(S)-1作为合成右美沙芬的中间体;B:环己胺氧化酶CHAO Y321I或亚胺还原酶IR40催化合成右美沙芬中间体(S)-1[14-15] |
| |
针对右美沙芬关键中间体(S)-1的不对称合成,我们开发了分别由环己胺氧化酶(cyclohexylamine oxidase, CHAO) 及亚胺还原酶(imine reductase, IR) 催化的两条生物合成路线(图 1B)。对来源于氧化短杆菌(Brevibacterium oxidans IH-35A) 的环己胺氧化酶BoCHAO进行研究发现野生型BoCHAO对(R)-1无催化活性,随后以BoCHAO为模板,进行结构指导的酶分子改造。通过筛选影响底物进出及结合的氨基酸残基获得突变体Y321I,该突变体对(R)-1的kcat为13.98 min–1,实现了催化效率为390 L/(mmol·min) 的明显提升。突变体Y321I与化学还原剂氨硼烷结合进行去消旋化反应,合成了(S)-1,其ee值和分离收率分别为98%和76%[14]。在去消旋化反应中使用的氨硼烷试剂在工业生产中存在安全隐患,为了能够使(S)-1a的制备更绿色,我们通过筛选发现来源于解淀粉橙色粘细菌(Sandaracinus amylolyticus) 的亚胺还原酶IR40催化亚胺底物可获得目标构型产物。以IR40与葡萄糖脱氢酶(glucose dehydrogenase, GDH) 共表达获得的菌株为催化剂,产物(S)-1的ee值和分离收率分别为98%和74%[15]。
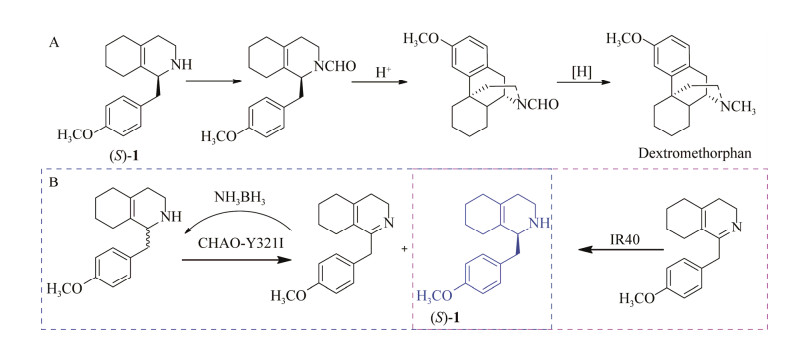
1.2 治疗失眠的药物苏沃雷生关键手性胺中间体的合成失眠是最常见的一种睡眠障碍,在人群中发病率高,超过3亿中国人有睡眠障碍。苏沃雷生(suvorexant) 于2014年获得批准在美国上市,同年在日本上市。苏沃雷生是全球首个以食欲素受体为作用位点的失眠治疗药物,用于治疗入睡及睡眠困难(失眠症)[16-17]。苏沃雷生的关键结构单元是一个手性1, 4-二氮䓬环((R)-2, 图 2),目前已报道的合成方法有手性拆分[18]、钌配合物催化分子内不对称还原胺化[19]及转氨酶催化分子内胺化闭环[20]等,但分别存在理论收率低、重金属残留及副产物多等问题,因此其高效不对称合成仍然具有重要应用价值。通过对苏沃雷生的逆合成分析发现氨基酮底物分子内的不对称还原胺化可实现苏沃雷生中间体的高效合成,形成手性胺的同时完成了1, 4-二氮䓬环的构建(图 2)。
|
|
| 图 2 分子内还原胺化合成苏沃雷生的逆合成路线 Fig. 2 Retrosynthetic route to suvorexant through intramolecular reductive amination. |
| |
利用前体氨基酮底物4-[(2-氨基乙基)(5-氯-2-苯并恶唑基)氨基]-2-丁酮甲磺酸盐筛选发现来源于硕大利什曼原虫(Leishmania major) 的亚胺还原酶IR1可合成(R)-2, ee值> 99%,但其催化效率仅有0.027 L/(mmol·s)。随后对IR1进行蛋白质工程改造获得了催化效率提高61倍的双突变体Y194F/D232H。该突变体在10 h内能够转化100 mmol/L底物,产物(R)-2的ee值及分离得率分别为 > 99%和81%[21]。亚胺还原酶催化的不对称还原胺化反应为苏沃雷生手性中间体的合成提供了一种环境友好型的高效合成方法。
1.3 烟草戒断剂关键手性胺中间体的合成使用诸如烟草或尼古丁之类的物质常常令使用者对这些物质成瘾或产生依赖,从而引起许多有害的后果,包括临床毒性、组织损伤和戒断症状以及维持社会和职业关系的能力受损。烟草戒断剂既可减轻烟瘾,又能明显地缓解戒断症状。(S)-去甲烟碱((S)-3) 是一种尼古丁代谢物和烟草生物碱,可作为烟草戒断剂使用于电子烟中[22],同时也具有神经药理学相关功能[23-24]和镇痛功能[25]等。化学法合成(S)-3需要多步反应,过程中使用的手性衍生剂或金属催化剂等容易造成环境污染,且产品光学纯度及收率不高,在实际大规模生产中有诸多限制[26-27]。
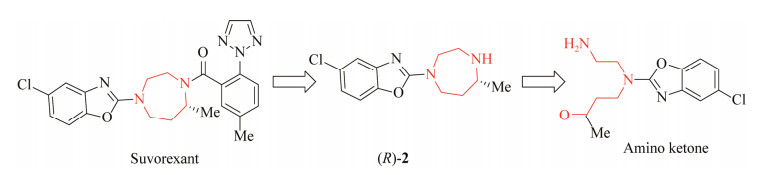
笔者团队筛选获得了来源于黄色粘球菌(Myxococcus fulvus) 的亚胺还原酶IR51,实现了由麦司明(myosmine) 不对称还原合成(S)-3,ee值> 99% (图 3)[28]。通过发酵条件优化实现了IR51与葡萄糖脱氢酶共表达菌株的高密度发酵,每升发酵液酶活可达15 000 U以上。该技术已转让相关企业并已建成年产200 t生产线,首次利用生物催化法在工业规模生产(S)-去甲烟碱。
|
|
| 图 3 亚胺还原酶催化不对称还原合成(S)-3 Fig. 3 Imine reductase-catalyzed enantioselective reduction to the intermediate of (S)-3. |
| |
手性胺作为重要的医药中间体,其不对称合成一直备受关注,随着生物催化技术的发展,转氨酶、亚胺还原酶、氨脱氢酶和水解酶等可以应用于手性胺的合成,每种转化方式都有其自身的特点,如有的酶底物谱较广泛,有的酶反应体系较为简单,因此在合成手性胺时可以针对胺的合成路线特点选择特定的生物催化剂。
2 手性醇类医药化学品的绿色生物合成在全球药品销售额TOP200的小分子药品,有52个药物分子具有手性醇结构(包含多糖结构),手性醇是重要的医药中间体和精细化工品,立体选择性羰基还原催化制备手性醇具有重要的研究与应用价值,受到国内外科学家和产业工程师的高度关注。羰基还原酶在温和的反应条件下实现羰基不对称还原,具有产物光学纯度高、生产成本低廉、环境友好等突出优势。因此,羰基还原酶作为催化剂制备手性醇的研究逐渐增多,并且在产业化应用上也有诸多报道。
2.1 妇女生殖健康药物左炔诺孕酮、依托孕烯关键手性醇中间体的合成左炔诺孕酮(levonorgestrel)、依托孕烯(gestodene) 是目前常用的应急性和长期避孕制剂。由于左炔诺孕酮的便利性、安全性和耐受性均好,且紧急避孕疗效高并不会受到共用药物的影响,加之用药前不需进行妊娠试验、产后哺乳或未哺乳妇女亦能使用,欧美各国以及中国都已批准左炔诺孕酮方案为非处方药物,年均销售额高达10亿美金以上。左炔诺孕酮、依托孕烯是全合成药物,工业化生产的合成路线基本定型,关键起始原料是氢化物(Z-2),而18甲缩合物(ethyl secodione) 去对称性还原制备(13R, 17S)-ethyl secol ((13R, 17S)-4) 是构建氢化物(Z-2) 13位手性中心的关键步骤,也是整条合成路线中唯一采用的生物催化步骤。该步骤以酵母作为催化剂,催化效率低,7 g/L的底物需要3 d才能完全转化[29] (图 4A)。
|
|
| 图 4 生物催化去对称还原环二酮及其应用 Fig. 4 Biocatalytic desymmetric reduction of cyclodiones and its applications. (A) Desymmetric carbonyl reduction of ethyl secodione as the key step in the production of levonorgestrel and gestodene. (B) The desymmetric reduction of 2, 2-disubstituted-1, 3-cyclodiones catalyzed by carbonyl reductase[31-32]. A:去对称羰基还原乙基西可二酮(ethyl secodione) 合成甾体药物左炔诺孕酮、依托孕烯;B:羰基还原酶催化2, 2-双取代-1, 3-环二酮的去对称还原[31-32] |
| |
针对这个转化难题,我们通过筛选确定以来源于罗尔斯通菌(Ralstonia sp.) 的羰基还原酶RasADH作为模板,采用结构指导的定向进化策略,经过数轮的改造筛选,获得了能够高效催化底物乙基西可二酮(ethyl secodione) 立体选择性还原的突变酶RasADH F12 (I91V/I187S/I188L/Q191N/F205A),该突变酶对底物的活力提高了183倍,目标产物(13R, 17S)-4的比例由37.3%提高至 > 99.5%,双羰基还原产物比例由18.8%降低至 < 0.1%[30]。该技术转让给相关企业,相较于现行的工业生产步骤效率提高了70倍,在国际上首次利用羰基还原酶实现(13R, 17S)-4的产业化生产。我们随后针对2, 2-双取代-1, 3-环二酮展开了进一步的研究,通过酶库的筛选、结构指导的酶改造实现了一系列的多手性中心2, 2-双取代-1, 3-环戊/己酮醇的精准构建(图 4B)[31-32]。
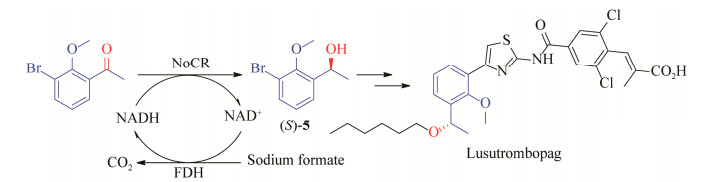
2.2 抗血小板减少症药物芦曲泊帕手性醇中间体的合成芦曲泊帕(lusutrombopag) 是人类血小板生成素受体激动剂,该药2015年在日本获得批准在选择进行侵入性手术的慢性肝脏疾病患者中使用,改善他们的血小板减少症病情;2018年获美国FDA批准,用于治疗患有血小板减少症的特定成人患者群体,这些患者同时患有慢性肝脏疾病,并有接受手术的计划。(S)-1-(2-甲氧基-3-溴苯基)乙醇((S)-5) 是芦曲泊帕的关键手性中间体[33],化学上主要采用(R)-2-甲基-CBS-恶唑硼烷催化硼烷四氢呋喃复合物不对称还原1-(2-甲氧基-3-溴苯基)乙酮的方法获得[34]。
通过广泛筛选羰基还原酶,我们获得了来源于新鞘氨醇菌(Novosphingobium sp. leaf2) 的羰基还原酶NoCR,该酶对苯环上有邻位和间位双取代酮1-(2-甲氧基-3-溴苯基)乙酮的比活性为15.4 U/mg,kcat/KM为11.3 L/(mmol·s)。将该酶与甲酸脱氢酶进行共表达后获得的菌株可以完全转化的底物浓度为200 g/L,产物(S)-5的ee值大于99% (图 5)[35]。
鲁索利替尼(Jakafi®) 是一种小分子Janus激酶抑制剂,用于治疗骨髓纤维化、抵抗形式的真性红细胞增多症和移植物抗宿主病。(S)-3-环戊基-3-羟基丙腈((S)-6) 是合成鲁索利替尼的关键手性中间体[36]。
筛选发现来源于帕拉伯克霍尔德菌(Paraburkholderia hospita) 的羰基还原酶PhADH催化3-环戊基-3-酮丙腈还原得到(S)-6,但ee值仅85%。对PhADH的突变筛选获得了双突变体H93C/A139L,该突变体的比活性提高了6.3倍,目标产物(S)-6的ee值从85%提高到 > 98%。H93C/A139L与葡萄糖脱氢酶共表达后获得的菌株在无助溶剂的反应条件下,可以将200 g/L β-环戊基-β-酮丙腈完全转化,以 > 98%的ee值和91%分离产率获得(S)-6。该突变酶对β-环己基-β-酮丙腈和β-苯基- β-酮丙腈同样具有在高浓度下完全转化的能力,以 > 99%的ee值获得相应手性醇(图 6)[37]。
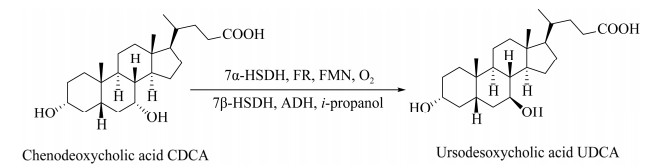
熊去氧胆酸可用于治疗胆固醇结石、慢性胆汁淤积性疾病、脂肪性肝病、病毒性肝炎等,全球需求量年均1 000 t左右。目前熊去氧胆酸制备工艺是以鹅去氧胆酸为底物,通过化学催化的氧化和还原反应实现,在氧化步骤用到N-溴代丁二酰亚胺,在还原步骤则要用到兰尼镍或金属钯催化,转化工艺后处理成本较高,对环境压力大。开发一条绿色可持续的制备熊去氧胆酸的工艺是非常迫切的。
基于羰基还原酶可逆性、专一性的催化特点,我们利用7α-羟基甾体脱氢酶与7β-羟基甾体脱氢酶作为催化剂,由鹅去氧胆酸(chenodeoxycholic acid, CDCA) 制备熊去氧胆酸(ursodesoxycholic acid, UDCA)。在转化过程中首次将黄素还原酶与核黄素应用到7α-羟基甾体脱氢酶对鹅去氧胆酸的氧化步骤中,实现氧化态辅酶的再生,共底物核黄素使用量仅为催化剂量,大大降低了鹅去氧胆酸的氧化转化成本。与7β-羟基甾体脱氢酶耦合实现了熊去氧胆酸的制备(图 7),建立的“一锅”多酶法生物合成熊去氧胆酸新工艺,已经转让给相关企业,实现了吨级规模生产,底物鹅去氧胆酸浓度不低于20 g/L,转化率大于95%,产物熊去氧胆酸的粗分离产率为95%,最终产物收率 > 90%,产品光学纯度 > 99%[38]。
2.5 重要香料二氢香芹醇的“一锅”多酶法催化合成二氢香芹醇(dihydrocarveol) 是一种香料,可以调制薄荷香型香精用于食品;可以调制留兰香型香精用于化妆品;也用作其他香料原料。目前市场上是以消旋体或者是非对映异构体混合物的形式出售。光学纯的二氢香芹醇则是贵重香料,也可作为手性原料用于合成生物活性的复杂分子。还原(R) /(S)-香芹酮(carvone) 可制备手性二氢香芹醇,单还原反应没有优异的区域选择性、化学选择性和非对映选择性的话,可能有17种产物生成[39]。
我们利用生物催化剂独特的专一性,将烯还原酶与羰基还原酶串联,利用葡萄糖脱氢酶/葡萄糖再生还原态辅酶NAD(P)H,分别以“一锅分步法”依次实现了(R) 和(S)-香芹酮的C=C、C=O双键的还原,获得了光学纯的二氢香芹醇((1S, 2R, 5R)-7和(1S, 2R, 5S)-8) (图 8)[40]。
(R)-和(S)-2-羟基丁酸是合成抗癫痫药物左乙拉西坦和布里瓦拉西坦的关键前体[41],还可广泛应用于生物可降解材料,具有很好的热性能和材料性能[42-43]。合成手性2-羟基丁酸包括传统化学方法[44-45]、酶动力学拆分[46-47]和2-酮丁酸的不对称生物还原[48-49]。
利用L-苏氨酸脱氨酶(L-threonine deaminase, TD)、甲酸脱氢酶(formate dehydrogenase, FDH) 和来自楔形谷杆菌(Oryctolagus cuniculus) 的L-乳酸脱氢酶(L-lactate dehydrogenase, L-LDH) 或来自表皮葡萄球菌(Staphylococcus epidermidis) ATCC 12228的D-乳酸脱氢酶(D-lactate dehydrogenase, D-LDH) 的三酶共表达菌,可将750 mmol/L的L-苏氨酸完全转化为(R)-或(S)-2-羟基丁酸,产物ee值> 99% (图 9)[50]。
手性醇作为重要的医药中间体,其手性合成一直备受关注,随着生物催化技术的发展,手性醇的获得方式由之前应用较广的脂肪酶拓展到了羰基还原酶(醇脱氢酶),随着蛋白质工程的发展,羰基还原酶研究的底物谱范围从简单酮到复杂酮都有报道,并且转化的底物浓度均较高(> 100 g/L),因此在合成手性醇时,羰基还原酶已经进入了产业化路线的设计中,并且体现出其优异的特性,逐渐成为精细化工行业的首选技术方法。
3 手性氨基酸类医药中间体的绿色生物合成手性氨基酸不仅在生命体代谢中具有重要作用,其在药物合成中也至关重要。在全球药品销售额TOP200的小分子药品,有38个药物分子具有手性α/β或γ-氨基酸结构(不包含10个由氨基酸构成的多肽药物分子),因此手性氨基酸的合成是生物催化的重要研究内容。本节将针对含有手性氨基酸药物分子的生物合成展开介绍,对多肽药物分子将不做介绍。
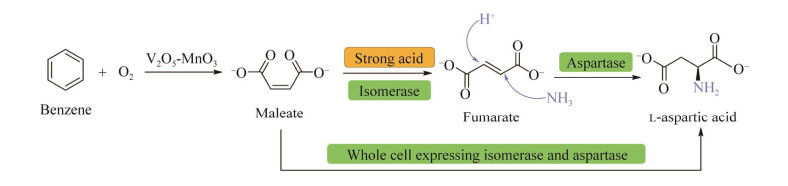
3.1 天然α-氨基酸——L-天冬氨酸的“一锅”多酶催化合成L-天冬氨酸又称L-氨基丁二酸,主要用于食品添加剂、阿巴斯甜、聚天冬氨酸、新药合成等领域,我国L-天冬氨酸产量达到6.5万t以上,占全球产量的65%以上。L-天冬氨酸传统生产工艺是裂解苯获得顺丁烯二酸(马来酸, maleate) 在强酸性条件(pH 1) 催化生成反丁烯二酸(富马酸, fumarate)。L-天冬氨酸氨基裂解酶催化氨与富马酸反应转化生成L-天冬氨酸铵,酸中和后分离纯化得到L-天冬氨酸。该工艺路线存在原料纯度要求高、分离纯化步骤多、能耗高和环境污染大等问题,成本和污染问题极大地限制了L-天冬氨酸的市场应用和价值的发挥(图 10)。
我们通过新酶设计、多酶级联催化及代谢通路改造相结合的方式,开发出第3代L-天冬氨酸绿色新生产工艺,该工艺利用异构酶和L-天冬氨酸氨基裂解酶共表达菌作为催化剂,新工艺设备匹配性高,不需要对现有的设备条件进行改造,生产过程中直接省去反丁烯二酸分离环节,极大简化生产工艺流程[51]。L-天冬氨酸的工业生产过程中节约50%生产用水、减少70%能源消耗、降低70%人工消耗并且消除强酸性废水排放(图 10),响应国家碳达峰碳中和政策。在合作企业建成年产万吨级规模生产线。产品各项指标全部符合L-天冬氨酸行业标准(QB/T 1118—1991, https://www.sdtdata.com/fx/fmrule/tsLibCard.doView),产品投放市场,获得下游用户高度认可。
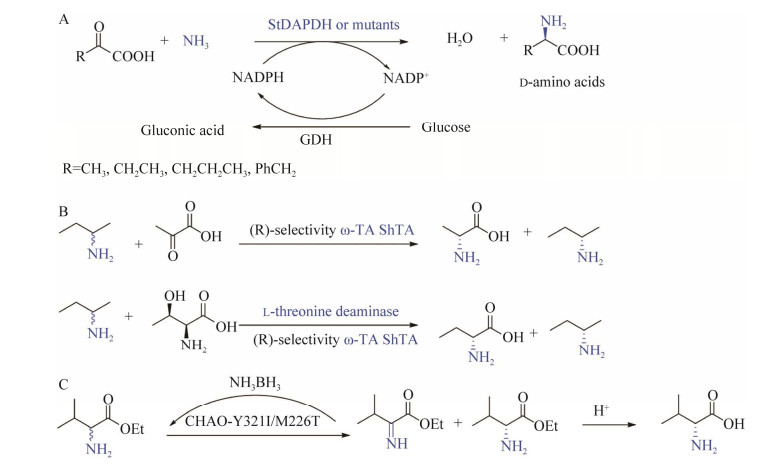
3.2 非天然α-氨基酸的生物催化合成 3.2.1 D-氨基酸的酶法合成D-氨基酸不仅在药物(医药和农药)、食品、化妆品、饲料等合成方面具有广泛的用途,而且在微生物、植物、动物以及人类体内具有多种生理功能[52-53]。能够合成D-氨基酸的酶有很多种,有转氨酶、环己胺氧化酶、D-氨基酸脱氢酶等,其中D-氨基酸脱氢酶直接催化游离氨与酮酸反应生成D-氨基酸的反应具有原子经济性和温和反应条件的优势,吸引了科研工作者和产业界的兴趣。
我们发现来源于嗜热梭菌(Symbiobacterium thermophilum) IAM14863的meso-二氨基庚二酸脱氢酶(diaminopimelic acid dehydrogenase, DAPDH) StDAPDH可以直接催化多种2-酮酸还原氨化生成D-氨基酸,该酶的热稳定性也是目前文献报道中最高的(图 11A)[54]。氨基酸序列和晶体结构研究表明StDAPDH独特的催化性质是其在氨基酸Met152的两边形成了两个底物进入通道,天然底物二氨基庚二酸只能通过其中一个较大的通道进入;而较小的底物,如D-丙氨酸和丙酮酸则可以通过较小的通道进出[55]。在此基础上,对底物结合位点周围的氨基酸残基分别进行定点饱和突变,获得多个立体选择性保持、活性提高的突变体,对丙酮酸、2-丁酮酸、2-戊酮酸、4-甲基-2-戊酮酸、苯基丙酮酸的酶活分别达到了32、32、30、9.91、2 U/mg,为野生酶的10、19、24、18和35.1倍。首次实现对3, 3-二甲基-2-丁酮酸的催化,产物D-叔亮氨酸ee值大于99%,虽然比活力只有0.006 U/mg[56-57]。将StDAPDH和甲酸脱氢酶固定化到树脂上得到共固定化酶,转化实验表明共固定化酶制备D-丙氨酸时显示出良好的立体选择性和可重复使用性,在重复使用12批次后保持80%的活性,前20批次反应的分离收率75%,ee值99.5%[58]。
|
|
| 图 11 酶法及酶-化学法合成D-氨基酸 Fig. 11 Enzymatic and chemo-enzymatic synthesis of D-amino acid. (A) D-amino acids synthesis by StDAPDH catalysis of keto acids and ammonia. (B) ω-transaminase catalyzed kinetic resolution to synthesis of D-alanine and D-2-aminobutanoic acid. (C) Deracemization of racemic ethyl valine by employing the CHAO mutant and NH3·BH3[56-60]. A:StDAPDH催化酮酸和氨合成D-氨基酸;B:ω-转氨酶催化动力学拆分合成D-丙氨酸和D-丁氨酸;C:CHAO和NH3·BH3共同作用实现外消旋缬氨酸乙酯的去消旋化[56-60] |
| |
ω-转氨酶同样可以用于合成D-氨基酸,以丙酮酸为氨基受体,利用来源于黄岛红球菌(Sinirhodobacter huangdaonensis) 的ω-转氨酶(ω-transaminase, TA) ShTA,可催化外消旋2-氨基丁烷(800 mmol/L) 的动力学拆分,分别以46%和90%的产率同时合成ee值为99%的(S)-2-氨基丁烷和D-丙氨酸(图 11B)。利用苏氨酸脱氨酶催化L-苏氨酸得到的2-丁酮酸为氨基受体,可催化外消旋2-氨基丁烷(400 mmol/L) 的动力学拆分,分别以45%和93%的产率同时制备ee值为99%的(S)-2-氨基丁烷和D-2-氨基丁酸(图 11B)[59]。CHAO突变体Y3211/M226T以外消旋缬氨酸乙酯或L-缬氨酸乙酯出发高效制备D-缬氨酸,ee值为 > 99%,收率为95%,突变体的催化效率比野生型CHAO高30倍(图 11C)。本方法可以利用廉价的消旋或L-氨基酸酯为底物,是合成D-缬氨酸的新方法[60]。
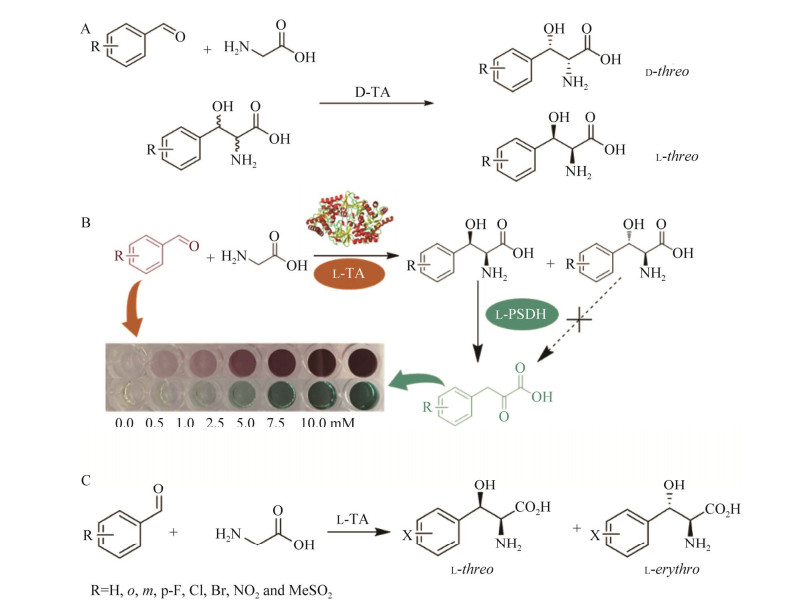
3.2.2 β-羟基-α-氨基酸的酶法合成手性β-羟基-α-氨基酸是许多药物的关键中间体,通过对羟基、氨基、羧基的进一步化学反应和修饰,能够合成氯霉素、甲砜霉素、万古霉素和氟苯尼考等一系列人用、兽用抗生素等。苏氨酸醛缩酶催化醛和甘氨酸不对称合成β-羟基-α-氨基酸,该反应一步生成2个手性中心,具有环境友好性,反应条件温和等优点。
D-苏氨酸醛缩酶(D-threonine aldolase, D-TA) 的底物特异性研究表明D-苏氨酸醛缩酶对多种芳香醛具有活性,吸电子取代基团的活性高于供电子取代基团的芳香醛。在最适反应条件下,2-氟苯甲醛和3-硝基苯甲醛与甘氨酸合成对应β-羟基-α-氨基酸获得了 > 90%的转化率和 > 90%的de值。在双相体系下,整细胞催化完全拆分1.0 mol/L D/L-threo-苯丝氨酸(D/L- threo-phenylserine, D/L-threo-PS) 和0.3 mol/L D/L-threo-4-甲砜基苯丝氨酸(D/L-threo-4- (methylsulfonyl) phenylserine, D/L-threo-MPS),获得了 > 90%的ee值(图 12A)[61]。L-苏氨酸醛缩酶(L-threonine aldolase, L-TA) 催化甘氨酸与醛缩合反应的产物α-碳的立体选择性能够得到绝对控制,但β-碳的立体选择性则往往较低,导致产物为混合物,限制了其在不对称合成中的应用。为了实现L-苏氨酸醛缩酶的改造,我们开发了该酶的高通量筛选方法。该高通量筛选方法是在反应后,偶联特异性催化threo-β-羟基-α-氨基酸生成酮酸的苯丝氨酸脱水酶,分步可视化检测苏氨酸醛缩酶催化的羟醛缩合的活性和立体选择性(图 12B)。随后我们构建L-苏氨酸醛缩酶的突变文库,通过开发的高通量筛选方法,在机理解析的基础上对酶分子进行针对性改造,获得对β-碳的立体选择性提高的突变体;利用获得的突变体实现β-羟基-α-氨基酸产物β-碳的立体选择性控制,尝试了利用L-苏氨酸醛缩酶为催化剂的β-羟基-α-氨基酸生物合成(图 12C)[62-63]。
|
|
| 图 12 苏氨酸醛缩酶的合成应用及酶改造的高通量筛选方法[61-63] Fig. 12 The synthetic application of threonine aldolase and high-throughput screening method for the selection of TA mutants[61-63]. (A) Asymmetric catalysis and kinetic resolution of D/L-threo-PS and MPS by D-threonine aldolase. (B) Reactions of colorimetric assays for benzaldehyde and phenylpyruvic acid. (C) Synthesis of β-hydroxy-α-amino acids employing L-threonine aldolase. A:D-苏氨酸醛缩酶催化不对称合成和动力学拆分制备D/L-threo-苯丝氨酸和对甲砜基苯丝氨酸;B:显色反应检测苯甲醛和苯基丙酮酸;C:L-苏氨酸醛缩酶催化合成β-羟基-α-氨基酸 |
| |
手性β-氨基酸不仅是多种天然化合物中的重要组成部分,也是许多合成药物和多肽的关键结构单元,在生命科学、药物开发和食品等领域具有重要的应用价值[64-68],因此,开发其高效、绿色的合成方法已是当前的研究热点。氨基酸脱氢酶(amino acid dehydrogenase, AADH) 催化的前手性酮酸与廉价的无机氨不对称还原胺化反应,因其副产物只有水,从原子经济性和对环境影响方面来看,都具有极大的优越性[69]。然而,与被广泛研究的α-AADHs相比,目前β-AADH家族中的已知成员只有L-赤式-3, 5-二氨基己酸脱氢酶(3, 5- diaminohexanoate dehydrogenase, 3, 5-DAHDH),且这类酶具有较严格的底物特异性,严重限制了其合成应用范围(图 13A)。在无3, 5-DAHDH这类酶相关晶体结构及同源建模比较困难的情况下,采用结构域逐点扫描的策略,对来源于嗜酸氨基泄殖腔单胞菌暂定种(Candidatus Cloacamonas acidaminovorans) 的3, 5-DAHDH (3, 5-DAHDHcca)[70]进行定向进化,获得了底物谱拓宽的2个突变体E310G/A314Y和E310S/A314N,实现了对非天然底物活性“从无到有”的突破,开辟了一条利用β-AADH不对称还原胺化β-酮酸制备手性β-氨基酸的新途径[71]。通过重新构建NADPH酰胺基团与附近残基的氢键相互作用和重塑底物口袋,在不牺牲对映选择性的前提下,获得了对多种脂肪族β-氨基酸活性提高110–800倍的优异突变体。利用这些突变体还原胺化相应β-酮酸,实现了(R)-β-高甲硫氨酸和(S)-3-氨基己酸的高效不对称合成,其ee值> 99%,分离产率可达86%–87%[72](图 13B)。通过对β-氨基酸脱氢酶的改造,开辟了一条绿色合成β-氨基酸的新途径,开创了利用氨基酸脱氢酶合成非天然氨基酸的新研究领域。
|
|
| 图 13 L-赤式-3, 5-二氨基己酸脱氢酶的合成应用[69-72] Fig. 13 Synthetic application of L-erythro-3, 5-diaminohexanoate dehydrogenase[69-72]. (A) L-erythro-3, 5-diaminohexanoate dehydrogenase catalyzed reaction. (B) Asymmetric synthesis of β-amino acids by multi-enzyme cascade strategy. A:L-赤式-3, 5-二氨基己酸脱氢酶催化的反应;B:多酶偶联反应不对称合成手性β-氨基酸 |
| |
γ-氨基丁酸是一种重要的中枢神经系统抑制性神经递质,其3位取代类似物可用于治疗与快速神经发放相关的某些病症。例如(R)-巴氯芬(Lioresal®) 是可用于缓解多发性硬化、脊髓空洞症、脊髓肿瘤、横贯性脊髓炎脊髓外伤运动神经元病及脑血管病、脑性瘫痪、脑膜炎颅脑外伤引起的骨骼肌痉挛;(S)-普瑞巴林(Lyrica®) 是首个被美国FDA批准用于缓解与治疗糖尿病周围神经病变和带状疱疹后神经痛的药物,普瑞巴林在全球超过130个国家上市,是市场上最畅销、重磅炸弹级镇痛药。至2019年6月,辉瑞制药有限公司的(S)-普瑞巴林及适用症的相关专利保护已全部到期,仿制药生产商的市场份额开始增加。(S)-普瑞巴林在工业上是采用辉瑞研发的脂肪酶拆分法进行合成,底物转化率为50%,需进行消旋化反应提高底物利用率[73-75]。γ-氨基丁酸前体的γ-氰基丁酸可以通过不对称催化水解前手性二腈化合物获得,与脂肪酶拆分最高仅为50%的方法相比,该方法转化率理论上实现底物的完全转化。
通过催化3-取代戊二腈筛选发现来源于蓝藻集胞藻(Synechocystis sp.) PCC6803的腈水解酶(nitrilase, NIT) SsNIT [76]具有较好的水解活性,利用结构指导的半理性改造策略,经过多轮改造筛选获得能够催化底物(4′-氯苯基) 戊二腈生成S构型手性氰基羧酸的突变体P194A/I201A/F202V,该突变体的时空产率达到野生型酶30倍以上,达到488 g/(Lžd),且选择性提高至99% (图 14A)。通过进一步模拟分析发现腈水解酶底物结合口袋发生了改变,被扩大的活性中心区域及活性口袋入口是突变体酶活性提高的原因[77-78]。随后我们通过底物结合口袋的镜像分析,将镜像位置的2个氨基酸残基的位置进行互换,实现了该酶对3-取代戊二腈化合物立体偏好性的翻转。经过改造筛选获得了能够高效、高选择性催化底物3-异丁基戊二腈的突变酶E8,该突变酶对底物的活力提高了32倍,并有效降低副产物氰基酰胺的含量由10%至1%以下,可高效催化400 mmol/L底物选择性水解,生成(R)-3-异丁基-4-氰基丁酸,分离产率97%,产物ee值大于99% (图 14B)[79-81]。该工作为腈水解酶及其他酶去对称化反应的立体选择性改造获得立体偏好性互补的酶催化剂提供了一种新的设计改造策略,也成功构建了(R)-巴氯芬及(S)-普瑞巴林关键中间体的酶法合成方法,为我国原料药的绿色高效制备作出了重要贡献。
|
|
| 图 14 腈水解酶的合成应用 Fig. 14 The synthetic application of nitrilase. (A) Hydrolysis of 3-(4′-chlorophenyl) glutaronitrile compounds by nitrilase and mutants. (B) Engineering nitrilase for the synthesis of stereoisomers. A:腈水解酶及其突变体催化水解3-(4′-氯苯基)戊二腈类化合物;B:腈水解酶突变改造合成不同构型产物 |
| |
天然、非天然氨基酸在手性药物合成中至关重要,随着生物催化剂底物谱拓展、性能强化、立体选择性调整,越来越多的氨基酸可以通过生物催化制备获得。
4 甾体药物关键中间体的绿色生物合成甾体药物广泛用于治疗风湿性关节炎、湿疹、支气管哮喘、过敏性休克、前列腺炎、爱迪森氏等疾病,亦可用于避孕、安胎、减轻女性更年期症状、手术麻醉等方面,以及预防冠心病、艾滋病、减肥等,是仅次于抗生素的第二大类药物[82-85]。在全球药品销售额TOP200的小分子药品,有9个药物分子具有甾体结构。
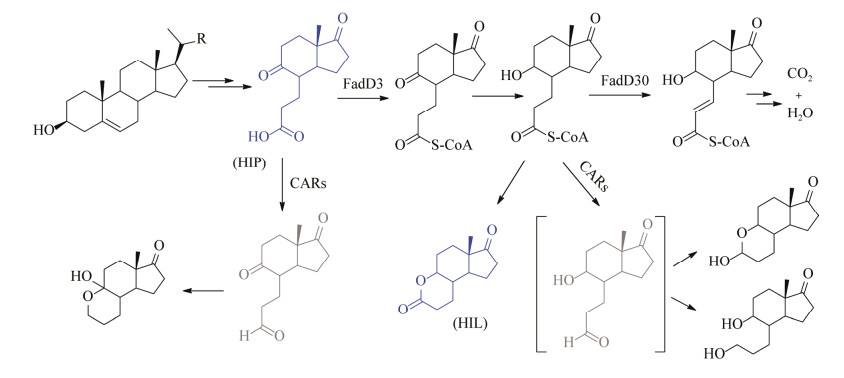
4.1 A环降解物(HIL和HIP) 的生物催化合成以偶发分支杆菌(Mycobacterium fortuitum ATCC 6841) 为母本构建了ΔfadD3 (Fad3为酰基辅酶A合酶) 和ΔfadE30 (Fad30为酰基辅酶A脱氢酶) 缺陷菌株,接着分别在ΔfadD3和ΔfadE30菌株中同时敲除2个羧酸还原酶(carboxylate reductase, CAR) 基因构建了新的菌株,分别代谢甾醇积累HIP和HIL,且未检测到其他副产物(图 15)。菌株ΔfadD3Δcar1, 2转化20 g/L植物甾醇可积累摩尔产率为88%的产物HIP,经重结晶后HIP分离产率为82%;相同条件下,菌株ΔfadE30Δcar1, 2生成产物HIL,摩尔产率为72%,分离产率为66%[86]。
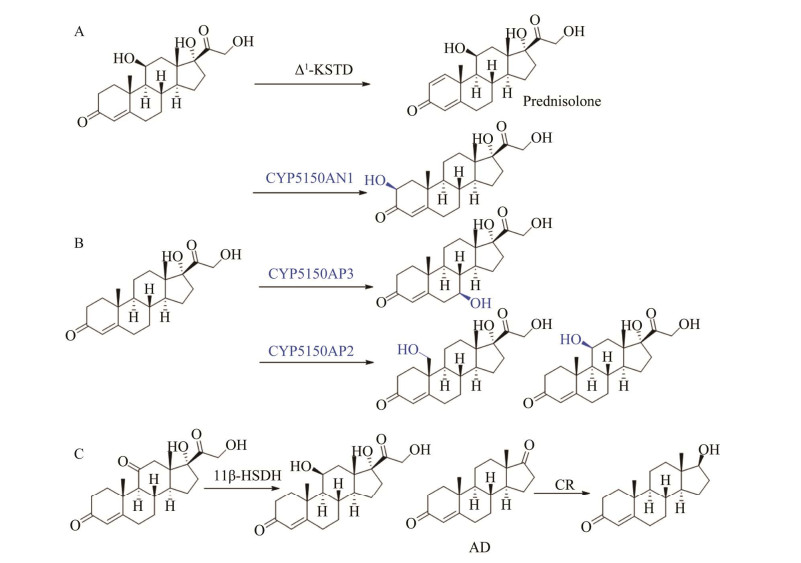
甾体1, 2-位脱氢反应是甾体药物合成中的重要反应之一,在1, 2-位引入C=C双键往往会增强现有的药用性能或显示出新的生物活性。3-甾酮化合物的Δ1-脱氢可以通过化学或生物催化方法来实现。化学脱氢法的局限性在于区域选择性低和使用大量有毒试剂、溶剂。3-甾酮-Δ1-脱氢酶(Δ1-KSTD) 能在温和条件催化3-甾酮化合物在母核A环C1, 2位脱氢,具有高选择性。我们通过对3-甾酮-Δ1-脱氢酶的挖掘和异源高效表达,实现了一系列3-甾酮化合物的Δ1-脱氢,其中PrKstD工程菌可在6 h内将80 g/L氢化可的松转化为泼尼松龙,转化率达到92.5% (图 16A)[87-88]。
|
|
| 图 16 酶催化的甾体官能化 Fig. 16 Enzyme-catalyzed functionalization of steroids. (A) The synthesis of prednisolone catalyzed by Δ1-KSTD. (B) The hydroxylation of cortodoxone catalyzed by CYP5150 family. (C) 11β-HSDH and carbonyl reductase mutants catalyzed the carbonyl reduction. A:Δ1-KSTD催化可的松合成泼尼松龙;B:CYP5150家族催化可托多松的羟基化反应;C:11β-羟基甾体脱氢酶及羰基还原酶突变体催化甾体的羰基还原 |
| |
19-去甲甾体是广泛存在于许多甾体药物和天然甾体产品中的核心结构,如炔诺酮(norethisterone)[89]、替勃龙(tibolone)、诺龙苯丙酸酯(durabolin)[90]、米非司酮(miferpristone)[91]等。C19位是一个特殊的角甲基结构,在该位置上羟基的引入是合成19-去甲基甾体化合物的关键环节[92]。水稻纹枯病菌(Thanatephorus cucumeris) 被发现可催化可托多松的羟基化反应生成19-羟基可托多松、11β-羟基可托多松和7β-羟基可托多松[93]。对T. cucumeris转录组数据分析后,利用酵母表达该菌株中的P450候选基因,通过转化实验验证功能。结果表明,CYP5150AP2可以实现对可托多松C19位和C11位的羟基化反应[94],CYP5150AN1和CYP5150AP3同时也从该菌中首次发掘并鉴定了7β-羟化酶和2β-羟化酶(图 16B)[95]。
4.4 羰基还原制备甾体药物中间体在甾体制药体系中,常常还会涉及在甾核C3、C11、C17等位置进行羰基与羟基的转换反应。由于甾核羟基具有特定的立体构型,因此羰基与羟基间的转换往往需保持高度的区域和立体选择性。对来源于豚鼠的11β-羟基甾体脱氢酶(11β-HSDH) 进行改造,有效地提高了该膜蛋白的可溶表达。通过与葡萄糖脱氢酶共表达构建的菌株实现了氢化可的松高选择性的生物合成(图 16C)[96]。对羰基还原酶RasADH进行定点饱和突变,筛选得到突变体RasADH F205I,相较于野生酶,其催化效率提高了6−623倍。RasADH F205I能催化一系列不同的17β-羟基甾体化合物的制备合成,在6 h内最高转化底物浓度为30 g/L,分离产率为72%–96%,de值> 99%,对底物雄烯二酮(AD) 的时空产率达到39.7 g/(L·d),远高于文献[97]报道水平[98](图 16C)。
4.5 以葡萄糖为唯一碳源从头合成薯蓣皂素薯蓣皂素是利用半合成方式生产甾体激素类药物(如可的松、避孕药和合成代谢药等) 的重要基础原料[99-100],因此被医药学家誉为“药用黄金”[101]。本研究所张学礼研究团队通过完善薯蓣皂素异源合成的功能模块、利用代谢工程策略提高元件与模块、途径与细胞之间的适配性、整合组学及细胞生理学分析将酿酒酵母薯蓣皂素人工细胞优化为一株全新的薯蓣皂素细胞工厂。该细胞工厂以葡萄糖为唯一碳源通过288 h的高密度分批补料发酵最终可产薯蓣皂素2.03 g/L,这是目前报道的利用微生物从头合成薯蓣皂素的最高产量[102]。
植物甾醇是食用油生产过程的副产物,以全球大豆产量3.3亿t/年计,仅大豆来源的植物甾醇即高达33–100万t;此外还有造纸工业等产生的难以计算的木甾醇等甾醇资源。植物甾醇已经是当前合成甾体类药物的重要前体。近几十年来,利用微生物降解甾醇是日本和西方国家生产甾体药物关键中间体的主要方法。植物甾醇资源的全面开发及高值化利用,有利于保证甾体医药市场的稳定,也有利于提高大豆等生物质资源的效益,进一步分摊关系民众日常生活的食用油等的成本。为了避免甾体药物的来源单一性,保持甾体药物价格的稳定,薯蓣皂素作为甾体的合成原料仍然需要保持和发展。
5 芳香族医药化学品的绿色生物合成芳香族化学品广泛应用于溶剂、塑料、纤维、香精香料、医药、食品等领域,市场规模高达上千亿美元,其中仅香精香料市场规模超400亿美元。目前芳香族化学品主要来自依赖化石资源的三苯工业。该生产工艺面临环境污染严重、发展不可持续的问题。近年来,随着合成生物学的发展、全球可持续发展观念的普及以及国家政策的扶持,使得利用微生物合成技术生产芳香族化学品逐渐成为一种有效的替代生产方式。
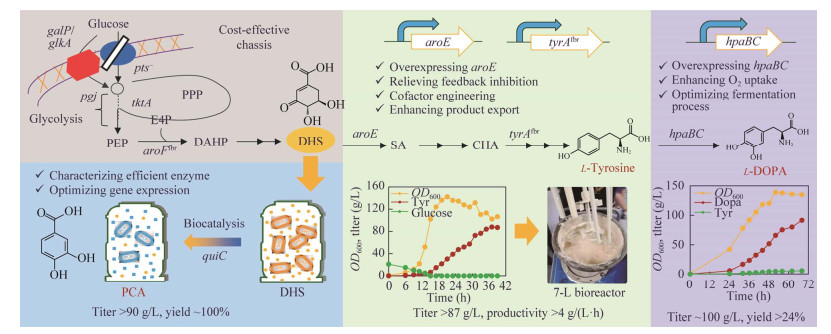
微生物细胞虽然可以自发合成各种芳香族化合物,但是受到细胞定向合成能力弱、物质流和能量流损失严重、合成模块不匹配、抗逆性低、产物细胞毒性强等因素的制约,导致产物滴度较低,难以满足工业化生产需求。为了解决上述关键科学问题,本研究所研究团队通过内源途径设计重构、异源途径整合适配、多位点基因迭代组合调控、实验室适应性进化和高通量筛选等多种研究技术和手段,创制了一系列具有自主知识产权的微生物细胞工厂,例如生产抗氧化剂3-脱氢莽草酸[103-105]、关键医药中间体原儿茶酸[106-107]、食品添加剂天然氨基酸L-酪氨酸[108]、治疗帕金森病特效药左旋多巴[109]等的细胞工厂(图 17)。上述芳香族化学品的产量、转化率以及生产强度等技术指标均达到国际领先水平,生产成本与化工路线相比有显著的环保优势和经济优势,“三废”排放减少30%–70%,生产成本降低60%以上。这些菌种项目转让给相关企业,多条百吨级产业化生产线建设正在推进中,投产后将形成良好的经济和社会效益,有助于推动我国芳香族化学品产业结构升级。
|
|
| 图 17 大肠杆菌细胞工厂绿色高效合成高附加值芳香族医药化合物 Fig. 17 Green and efficient synthesis of high value-added pharmaceutical aromatic compounds by Escherichia coli cell factory. |
| |
手性化学品生物制造已成为世界各国优先发展的科技和产业重点,通过生物催化方法制造手性医药化学品受到普遍关注,并已成为发达国家重要的科技和产业发展战略。分子生物学技术的发展使得人们可以根据不同的化学反应设计所需的酶催化剂,改造的生物酶在区域选择性、对映选择性控制以及不活泼位点的活化性能方面显著提升。未来手性医药化学品的生物制造,一方面是如何运用分子生物学和基因工程技术对生物催化剂性能进一步提升,实现生物催化剂的快速商品化和工具化;另一方面是如何将生物制造与现有的传统化学制造紧密结合,实现手性化学品的绿色制造和产业的可持续发展。
| [1] |
Strohmeier GA, Pichler H, May O, et al. Application of designed enzymes in organic synthesis. Chem Rev, 2011, 111(7): 4141-4164. DOI:10.1021/cr100386u
|
| [2] |
Albarrán-Velo J, González-Martínez D, Gotor-Fernández V. Stereoselective biocatalysis: a mature technology for the asymmetric synthesis of pharmaceutical building blocks. Biocatal Biotransform, 2018, 36(2): 102-130. DOI:10.1080/10242422.2017.1340457
|
| [3] |
Rocha RA, Speight RE, Scott C. Engineering enzyme properties for improved biocatalytic processes in batch and continuous flow. Org Process Res Dev, 2022, 26(7): 1914-1924. DOI:10.1021/acs.oprd.1c00424
|
| [4] |
Kar S, Sanderson H, Roy K, et al. Green chemistry in the synthesis of pharmaceuticals. Chem Rev, 2022, 122(3): 3637-3710. DOI:10.1021/acs.chemrev.1c00631
|
| [5] |
Hollmann F, Opperman DJ, Paul CE. Biocatalytic reduction reactions from a chemist's perspective. Angew Chem Int Ed Engl, 2021, 60(11): 5644-5665. DOI:10.1002/anie.202001876
|
| [6] |
Taylor CP, Traynelis SF, Siffert J, et al. Pharmacology of dextromethorphan: relevance to dextromethorphan/ quinidine (Nuedexta®) clinical use. Pharmacol Ther, 2016, 164: 170-182. DOI:10.1016/j.pharmthera.2016.04.010
|
| [7] |
Nguyen L, Thomas KL, Lucke-Wold BP, et al. Dextromethorphan: an update on its utility for neurological and neuropsychiatric disorders. Pharmacol Ther, 2016, 159: 1-22. DOI:10.1016/j.pharmthera.2016.01.016
|
| [8] |
Kumaraguru T, Fadnavis NW. An improved process for the preparation of (+)-3-methoxy-N-formylmorphinan. Org Process Res Dev, 2014, 18(1): 174-178. DOI:10.1021/op400309q
|
| [9] |
Meyers AI, Bailey TR. An asymmetric synthesis of (+)-morphinans in high enantiomeric purity. J Org Chem, 1986, 51(6): 872-875. DOI:10.1021/jo00356a023
|
| [10] |
Brossi A, Schnider O, Hydroxy-morphinane. 9. mitteilung. versuche zur racemisierung optisch aktiver 1-(p-hydroxybenzyl)-1, 2, 3, 4, 5, 6, 7, 8-octahydro-isochinoline. Helvetica Chimica Acta, 1956, 39(5): 1376-1386. DOI:10.1002/hlca.19560390525
|
| [11] |
Murakami M, Kawahara S, Inukai N, et al. Studies on morphinan derivatives. Ⅱ. The synthesis of d-3-methyl-N-methylmorphinan, a new antitussive. Chem Pharm Bull (Tokyo), 1972, 20(8): 1706-1710. DOI:10.1248/cpb.20.1706
|
| [12] |
Kitamura M, Hsiao Y, Noyori R, et al. General asymmetric synthesis of benzomorphans and morphinans via enantioselective hydrogenation. Tetrahedron Lett, 1987, 28(41): 4829-4832. DOI:10.1016/S0040-4039(00)96636-X
|
| [13] |
Broger EA, Burkart W, Hennig M, et al. New amidophosphine-phosphinites (tLANOPs) as chiral ligands for asymmetric hydrogenation reactions. Tetrahedron Asymmetry, 1998, 9(22): 4043-4054. DOI:10.1016/S0957-4166(98)00423-6
|
| [14] |
Li GY, Yao PY, Cong PQ, et al. New recombinant cyclohexylamine oxidase variants for deracemization of secondary amines by orthogonally assaying designed mutants with structurally diverse substrates. Sci Reports, 2016, 6: 24973.
|
| [15] |
Yao PY, Xu ZF, Yu SS, et al. Imine reductase-catalyzed enantioselective reduction of bulky α, β-unsaturated imines en route to a pharmaceutically important morphinan skeleton. Adv Synth Catal, 2019, 361(3): 556-561. DOI:10.1002/adsc.201801326
|
| [16] |
Yang LPH. Suvorexant: first global approval. Drugs, 2014, 74(15): 1817-1822. DOI:10.1007/s40265-014-0294-5
|
| [17] |
Cox CD, Breslin MJ, Whitman DB, et al. Discovery of the dual orexin receptor antagonist [(7R)-4-(5-chloro-1, 3-benzoxazol-2-yl)-7-methyl-1, 4-diazepan-1-yl][5-methyl- 2-(2H-1, 2, 3-triazol-2-yl)phenyl]methanone (MK-4305) for the treatment of insomnia. J Med Chem, 2010, 53(14): 5320-5332. DOI:10.1021/jm100541c
|
| [18] |
Baxter CA, Cleator E, Brands KMJ, et al. The first large-scale synthesis of MK-4305: a dual orexin receptor antagonist for the treatment of sleep disorder. Org Process Res Dev, 2011, 15(2): 367-375. DOI:10.1021/op1002853
|
| [19] |
Strotman NA, Baxter CA, Brands KMJ, et al. Reaction development and mechanistic study of a ruthenium catalyzed intramolecular asymmetric reductive amination en route to the dual orexin inhibitor suvorexant (MK-4305). J Am Chem Soc, 2011, 133(21): 8362-8371. DOI:10.1021/ja202358f
|
| [20] |
Mangion IK, Sherry BD, Yin JJ, et al. Enantioselective synthesis of a dual orexin receptor antagonist. Org Lett, 2012, 14(13): 3458-3461. DOI:10.1021/ol3014123
|
| [21] |
Xu Z, Yao P, Sheng X, et al. Biocatalytic access to 1, 4-diazepanes via imine reductase-catalyzed intramolecular asymmetric reductive amination. ACS Catalysis, 2020, 10(15): 8780-8787. DOI:10.1021/acscatal.0c02400
|
| [22] |
Stairs DJ, Neugebauer NM, Wei X, et al. Effects of nornicotine enantiomers on intravenous S(−)-nicotine self-administration and cardiovascular function in rats. Psychopharmacology, 2007, 190(2): 145-155.
|
| [23] |
Dwoskin LP, Teng LH, Crooks PA. Nornicotine, a nicotine metabolite and tobacco alkaloid: desensitization of nicotinic receptor-stimulated dopamine release from rat striatum. Eur J Pharmacol, 2001, 428(1): 69-79. DOI:10.1016/S0014-2999(01)01283-3
|
| [24] |
Middleton LS, Crooks PA, Wedlund PJ, et al. Nornicotine inhibition of dopamine transporter function in striatum via nicotinic receptor activation. Synapse, 2007, 61(3): 157-165. DOI:10.1002/syn.20351
|
| [25] |
Holtman JR Jr, Crooks PA, Johnson-Hardy JK, et al. The analgesic and toxic effects of nornicotine enantiomers alone and in interaction with morphine in rodent models of acute and persistent pain. Pharmacol Biochem Behav, 2010, 94(3): 352-362. DOI:10.1016/j.pbb.2009.09.017
|
| [26] |
Swango JH, Bhatti BS, Qureshi MM, et al. A novel enantioselective synthesis of (S)-(−)- and (R)-(+)-nornicotine via alkylation of a chiral 2-hydroxy-3-pinanone ketimine template. Chirality, 1999, 11(4): 316-318. DOI:10.1002/(SICI)1520-636X(1999)11:4<316::AID-CHIR9>3.0.CO;2-D
|
| [27] |
Wagner FF, Comins DL. Recent advances in the synthesis of nicotine and its derivatives. Tetrahedron, 2007, 63(34): 8065-8082. DOI:10.1016/j.tet.2007.04.100
|
| [28] |
李键煚, 刘祥涛, 徐泽菲, 等. 亚胺还原酶突变体及其应用: 中国, 202111270046.1, 2021-10-29. Li JJ, Liu XT, Xu ZF, et al. The imine reduct reductase mutants and their applications: CN, 202111270046.1, 2021-10-29 (in Chinese). |
| [29] |
周维善, 庄治平, 王锤麒. 对称二酮类化合物的微生物不对称还原——Ⅱ. 2-烷基-2-(3′-乙二醇缩酮丁基)-1, 3-环戊二酮的不对称还原. 化学学报, 1982, 40(7): 666-669. Zhou WS, Zhuang ZP, Wang ZQ. Asymmetrical reduction of the symmetrical diketones by micro- organism—ⅱ. asymmetrical reduction of 2-alkyl-2- (3′-ethyleneglycol ketal-butyl)-1, 3-cyclopentanedione. Acta Chimica Sin, 1982, 40(7): 666-669 (in Chinese). |
| [30] |
Chen X, Zhang H, Maria-Solano MA, et al. Efficient reductive desymmetrization of bulky 1, 3-cyclodiketones enabled by structure-guided directed evolution of a carbonyl reductase. Nat Catal, 2019, 2(10): 931-941. DOI:10.1038/s41929-019-0347-y
|
| [31] |
Li J, Feng JH, Chen X, et al. Structure-guided directed evolution of a carbonyl reductase enables the stereoselective synthesis of (2S, 3S)-2, 2-disubstituted- 3-hydroxycyclopentanones via desymmetric reduction. Org Lett, 2020, 22(9): 3444-3448. DOI:10.1021/acs.orglett.0c00892
|
| [32] |
Zhu LY, Cui YF, Chen X, et al. Synthesis of single stereoisomers of 2, 2-disubstituted 3-hydroxycyclohexane-1-ones via enzymatic desymmetric reduction of the 1, 3-cyclohexanediones. Green Synth Catal, 2021, 2(3): 320-323. DOI:10.1016/j.gresc.2021.04.009
|
| [33] |
Shirley M, McCafferty EH, Blair HA. Lusutrombopag: a review in thrombocytopenia in patients with chronic liver disease prior to a scheduled procedure. Drugs, 2019, 79(15): 1689-1695. DOI:10.1007/s40265-019-01197-8
|
| [34] |
Takayama M, Kurose N. Pharmaceutical composition containing optically active compound having thrombopoietin receptor agonist activity and intermediate thereof. CA2694567, 2008-07-29.
|
| [35] |
Cui YF, Ji YY, Chen X, et al. Efficient enzymatic synthesis of (S)-1-(3′-bromo-2′-methoxyphenyl) ethanol, the key building block of lusutrombopag. Green Synth Catal, 2022, Doi: .j.gresc.2022.06.010
|
| [36] |
王永, 刘利刚, 项杰, 等. 一种鲁索利替尼中间体的合成方法: 中国, 201410708821.0, 2014-11-28. Wang Y, Liu LG, Xiang J, et al. A method for the synthesis of the intermeediate of ruxolitinib: CN, 201410708821.0, 2014-11-28 (in Chinese). |
| [37] |
Cui YF, Zhu LY, Chen X, et al. Engineering a carbonyl reductase for scalable preparation of (S)-3-cyclopentyl-3-hydroxypropanenitrile, the key building block of ruxolitinib. ChemBioChem, 2022, 23(5): e202100589.
|
| [38] |
Chen X, Cui YF, Feng JH, et al. Flavin oxidoreductase-mediated regeneration of nicotinamide adenine dinucleotide with dioxygen and catalytic amount of flavin mononucleotide for one-pot multi-enzymatic preparation of ursodeoxycholic acid. Adv Synth Catal, 2019, 361(11): 2497-2504.
|
| [39] |
Dr RNP, Dr TOAP. Asymmetric catalysis by architectural and functional molecular engineering: practical chemo- and stereoselective hydrogenation of ketones. Angewandte Chemie Int Ed, 2001, 40(1): 40-73. DOI:10.1002/1521-3773(20010105)40:1<40::AID-ANIE40>3.0.CO;2-5
|
| [40] |
Chen X, Gao XZ, Wu QQ, et al. Synthesis of optically active dihydrocarveol via a stepwise or one-pot enzymatic reduction of (R)- and (S)-carvone. Tetrahedron Asymmetry, 2012, 23(10): 734-738. DOI:10.1016/j.tetasy.2012.05.019
|
| [41] |
Pàmies O, Bäckvall JE. Efficient lipase-catalyzed kinetic resolution and dynamic kinetic resolution of β-hydroxy nitriles. A route to useful precursors for γ-amino alcohols. Adv Synth Catal, 2001, 343(6/7): 726-731.
|
| [42] |
Tsuji H, Okumura A. Stereocomplex formation between enantiomeric substituted poly(lactide)s: blends of poly [(S)-2-hydroxybutyrate]and poly [(R)-2-hydroxybutyrate. Macromolecules, 2009, 42(19): 7263-7266. DOI:10.1021/ma9015483
|
| [43] |
Park SJ, Jang YA, Lee H, et al. Metabolic engineering of Ralstonia eutropha for the biosynthesis of 2-hydroxyacid-containing polyhydroxyalkanoates. Metab Eng, 2013, 20: 20-28. DOI:10.1016/j.ymben.2013.08.002
|
| [44] |
Shibuya K, Araki T, Koura M, et al. Novel synthesis of butyl (S)-2-hydroxybutanoate, the key intermediate of PPARα agonist (R)-K-13675 from butyl (2S, 3R)- epoxybutanoate and butyl (S)-2, 3-epoxypropanoate. Synthesis, 2013, 45(10): 1319-1324. DOI:10.1055/s-0032-1318507
|
| [45] |
Yamazaki Y, Araki T, Koura M, et al. A practical synthesis of the PPARα agonist, (R)-K-13675, starting from (S)-2-hydroxybutyrolactone. Tetrahedron, 2008, 64(35): 8155-8158. DOI:10.1016/j.tet.2008.06.049
|
| [46] |
Wang YJ, Lv M, Zhang YX, et al. Reconstruction of lactate utilization system in Pseudomonas putida KT2440: a novel biocatalyst for l-2-hydroxy-carboxylate production. Sci Rep, 2014, 4: 6939.
|
| [47] |
Gao C, Zhang W, Huang YL, et al. Efficient conversion of 1, 2-butanediol to (R)-2-hydroxybutyric acid using whole cells of Gluconobacter oxydans. Bioresour Technol, 2012, 115: 75-78. DOI:10.1016/j.biortech.2011.11.009
|
| [48] |
Jun CH, Sa YS, Gu SA, et al. Discovery and characterization of a thermostable d-lactate dehydrogenase from Lactobacillus jensenii through genome mining. Process Biochem, 2013, 48(1): 109-117. DOI:10.1016/j.procbio.2012.11.013
|
| [49] |
Zheng Z, Sheng B, Gao C, et al. Highly stereoselective biosynthesis of (R)-α-hydroxy carboxylic acids through rationally re-designed mutation of D-lactate dehydrogenase. Sci Reports, 2013, 3: 3401.
|
| [50] |
Yao PY, Cui YF, Yu SS, et al. Efficient biosynthesis of (R)- or (S)-2-hydroxybutyrate from L-threonine through a synthetic biology approach. Adv Synth Catal, 2016, 358(18): 2923-2928. DOI:10.1002/adsc.201600468
|
| [51] |
朱敦明, 吴洽庆, 李正华, 等. 一种新的顺丁烯二酸异构酶及应用: 中国, 2011100262975, 2011-01-15. Zhu DM, Wu QQ, Li ZH, et al. A novel maleic acid isomerase and its application: CN, 2011100262975, 2011-01-15 (in Chinese). |
| [52] |
Pollegioni L, Rosini E, Molla G. Advances in enzymatic synthesis of D-amino acids. Int J Mol Sci, 2020, 21(9): 3206. DOI:10.3390/ijms21093206
|
| [53] |
Gao XZ, Ma QY, Zhu HL. Distribution, industrial applications, and enzymatic synthesis of D-amino acids. Appl Microbiol Biotechnol, 2015, 99(8): 3341-3349. DOI:10.1007/s00253-015-6507-3
|
| [54] |
Gao XZ, Chen X, Liu WD, et al. A novel meso-diaminopimelate dehydrogenase from Symbiobacterium thermophilum: overexpression, characterization, and potential for D-amino acid synthesis. Appl Environ Microbiol, 2012, 78(24): 8595-8600. DOI:10.1128/AEM.02234-12
|
| [55] |
Liu WD, Li Z, Huang CH, et al. Structural and mutational studies on the unusual substrate specificity of meso-diaminopimelate dehydrogenase from Symbiobacterium thermophilum. ChemBioChem, 2014, 15(2): 217-222. DOI:10.1002/cbic.201300691
|
| [56] |
Cheng XK, Chen X, Feng JH, et al. Structure-guided engineering of meso-diaminopimelate dehydrogenase for enantioselective reductive amination of sterically bulky 2-keto acids. Catal Sci Technol, 2018, 8(19): 4994-5002. DOI:10.1039/C8CY01426D
|
| [57] |
Gao XZ, Huang F, Feng JH, et al. Engineering the meso-diaminopimelate dehydrogenase from Symbiobacterium thermophilum by site saturation mutagenesis for D-phenylalanine synthesis. Appl Environ Microbiol, 2013, 79(16): 5078-5081. DOI:10.1128/AEM.01049-13
|
| [58] |
Li JJ, Chen X, Cui YF, et al. Enzymatic synthesis of D-alanine from a renewable starting material by co-immobilized dehydrogenases. Process Biochem, 2018, 66: 126-132. DOI:10.1016/j.procbio.2017.12.008
|
| [59] |
Li JJ, Yu SS, Wang YG, et al. Simultaneous preparation of (S)-2-aminobutane and D-alanine or D-homoalanine via biocatalytic transamination at high substrate concentration. Org Process Res Dev, 2022, 26(7): 2013-2020. DOI:10.1021/acs.oprd.1c00408
|
| [60] |
Gong R, Yao PY, Chen X, et al. Accessing D-valine synthesis by improved variants of bacterial cyclohexylamine oxidase. ChemCatChem, 2018, 10(2): 387-390. DOI:10.1002/cctc.201701229
|
| [61] |
Chen QJ, Chen X, Cui YF, et al. A new D-threonine aldolase as a promising biocatalyst for highly stereoselective preparation of chiral aromatic β-hydroxy-α-amino acids. Catal Sci Technol, 2017, 7(24): 5964-5973. DOI:10.1039/C7CY01774J
|
| [62] |
Chen QJ, Chen X, Feng JH, et al. Improving and inverting cβ-stereoselectivity of threonine aldolase via substrate-binding-guided mutagenesis and a stepwise visual screening. ACS Catal, 2019, 9(5): 4462-4469. DOI:10.1021/acscatal.9b00859
|
| [63] |
Liu ZC, Chen X, Chen QJ, et al. Engineering of L-threonine aldolase for the preparation of 4-(methylsulfonyl) phenylserine, an important intermediate for the synthesis of florfenicol and thiamphenicol. Enzyme Microb Technol, 2020, 137: 109551. DOI:10.1016/j.enzmictec.2020.109551
|
| [64] |
Kudo F, Miyanaga A, Eguchi T. Biosynthesis of natural products containing β-amino acids. Nat Prod Rep, 2014, 31(8): 1056-1073. DOI:10.1039/C4NP00007B
|
| [65] |
Cabrele C, Martinek TA, Reiser O, et al. Peptides containing β-amino acid patterns: challenges and successes in medicinal chemistry. J Med Chem, 2014, 57(23): 9718-9739. DOI:10.1021/jm5010896
|
| [66] |
Kiss L, Mándity IM, Fülöp F. Highly functionalized cyclic β-amino acid moieties as promising scaffolds in peptide research and drug design. Amino Acids, 2017, 49(9): 1441-1455. DOI:10.1007/s00726-017-2439-9
|
| [67] |
Henninot A, Collins JC, Nuss JM. The current state of peptide drug discovery: back to the future. J Med Chem, 2018, 61(4): 1382-1414. DOI:10.1021/acs.jmedchem.7b00318
|
| [68] |
Katoh T, Sengoku T, Hirata K, et al. Ribosomal synthesis and de novo discovery of bioactive foldamer peptides containing cyclic β-amino acids. Nat Chem, 2020, 12(11): 1081-1088. DOI:10.1038/s41557-020-0525-1
|
| [69] |
Xue YP, Cao CH, Zheng YG. Enzymatic asymmetric synthesis of chiral amino acids. Chem Soc Rev, 2018, 47(4): 1516-1561. DOI:10.1039/C7CS00253J
|
| [70] |
Pelletier E, Kreimeyer A, Bocs S, et al. "Candidatus Cloacamonas acidaminovorans": genome sequence reconstruction provides a first glimpse of a new bacterial division. J Bacteriol, 2008, 190(7): 2572-2579. DOI:10.1128/JB.01248-07
|
| [71] |
Zhang DL, Chen X, Chi J, et al. Semi–rational engineering a carbonyl reductase for the enantioselective reduction of β-amino ketones. ACS Catal, 2015, 5(4): 2452-2457. DOI:10.1021/acscatal.5b00226
|
| [72] |
Liu N, Wu L, Feng JH, et al. Crystal structures and catalytic mechanism of L-erythro-3, 5-diaminohexanoate dehydrogenase and rational engineering for asymmetric synthesis of β-amino acids. Angew Chem Int Ed Engl, 2021, 60(18): 10203-10210. DOI:10.1002/anie.202017225
|
| [73] |
Martinez CA, Hu SH, Dumond Y, et al. Development of a chemoenzymatic manufacturing process for pregabalin. Org Process Res Dev, 2008, 12(3): 392-398. DOI:10.1021/op7002248
|
| [74] |
Jung JH, Yoon DH, Kang P, et al. CAL-B catalyzed desymmetrization of 3-alkylglutarate: "olefin effect" and asymmetric synthesis of pregabalin. Org Biomol Chem, 2013, 11(22): 3635-3641. DOI:10.1039/c3ob40311d
|
| [75] |
Zheng RC, Wang TZ, Fu DJ, et al. Biocatalytic synthesis of chiral intermediate of pregabalin with high substrate loading by a newly isolated Morgarella morganii ZJB-09203. Appl Microbiol Biotechnol, 2013, 97(11): 4839-4847. DOI:10.1007/s00253-013-4810-4
|
| [76] |
Duan YT, Yao PY, Ren J, et al. Biocatalytic desymmetrization of 3-substituted glutaronitriles by nitrilases. A convenient chemoenzymatic access to optically active (S)-pregabalin and (R)-baclofen. Sci China Chem, 2014, 57(8): 1164-1171. DOI:10.1007/s11426-014-5139-2
|
| [77] |
Yu SS, Yao PY, Li JL, et al. Improving the catalytic efficiency and stereoselectivity of a nitrilase from Synechocystis sp. PCC6803 by semi-rational engineering en route to chiral γ-amino acids. Catal Sci Technol, 2019, 9(6): 1504-1510. DOI:10.1039/C8CY02455C
|
| [78] |
于珊珊, 姚培圆, 冯进辉, 等. 腈基水解酶突变体及其在(S)-单腈单酸合成中的应用: 中国, 2018114659900, 2018-12-03. Yu SS, Yao PY, Feng JH, et al. Nitrilase mutant and application thereof in (S)-nitrilase single-acid synthesis: CN, 2018114659900, 2018-12-03 (in Chinese). |
| [79] |
Yu SS, Li JL, Yao PY, et al. Inverting the enantiopreference of nitrilase-catalyzed desymmetric hydrolysis of prochiral dinitriles by reshaping the binding pocket with a mirror-image strategy. Angew Chem Int Ed Engl, 2021, 60(7): 3679-3684. DOI:10.1002/anie.202012243
|
| [80] |
于珊珊, 姚培圆, 冯进辉, 等. 腈水解酶突变蛋白及其在催化合成(R) -3-取代-4-氰基丁酸类化合物中的应用: 中国, 2019106133546, 2019-07-09. Yu SS, Yao PY, Feng JH, et al. Nitrilase mutant protein and application thereof in catalytic synthesis of (R)-3-substituted-4-cyanobutyric acid compounds: CN, 2019106133546, 2019-07-09 (in Chinese). |
| [81] |
于珊珊, 姚培圆, 冯进辉, 等. 腈水解酶突变体及其应用: 中国, 2019111895573, 2019-11-28. Yu SS, Yao PY, Feng JH. et al. Nitrilase mutants and uses thereof: CN, 2019111895573, 2019-11-28 (in Chinese). |
| [82] |
Fernández-Cabezón L, Galán B, García JL. New insights on steroid biotechnology. Front Microbiol, 2018, 9: 958. DOI:10.3389/fmicb.2018.00958
|
| [83] |
Gupta P, Mahajan A. Sustainable approaches for steroid synthesis. Environ Chem Lett, 2019, 17(2): 879-895. DOI:10.1007/s10311-018-00845-x
|
| [84] |
Sprung CL, Goodman S, Weiss YG. Steroid therapy of septic shock. Crit Care Nurs Clin North Am, 2011, 23(1): 171-180. DOI:10.1016/j.ccell.2010.12.010
|
| [85] |
Tong WY, Dong X. Microbial biotransformation: recent developments on steroid drugs. Recent Pat Biotechnol, 2009, 3(2): 141-153. DOI:10.2174/187220809788700157
|
| [86] |
Liu N, Feng JH, Zhang R, et al. Efficient microbial synthesis of key steroidal intermediates from bio-renewable phytosterols by genetically modified Mycobacterium fortuitum strains. Green Chem, 2019, 21(15): 4076-4083. DOI:10.1039/C9GC01267B
|
| [87] |
Wang XJ, Feng JH, Zhang DL, et al. Characterization of new recombinant 3-ketosteroid-Δ1-dehydrogenases for the biotransformation of steroids. Appl Microbiol Biotechnol, 2017, 101(15): 6049-6060. DOI:10.1007/s00253-017-8378-2
|
| [88] |
Wang Y, Zhang R, Feng JH, et al. A new 3-ketosteroid-Δ1–dehydrogenase with high activity and broad substrate scope for efficient transformation of hydrocortisone at high substrate concentration. Microorganisms, 2022, 10(3): 508. DOI:10.3390/microorganisms10030508
|
| [89] |
Amsterdam A, Sasson R. The anti-inflammatory action of glucocorticoids is mediated by cell type specific regulation of apoptosis. Mol Cell Endocrinol, 2002, 189(1-2): 1-9. DOI:10.1016/S0303-7207(01)00722-5
|
| [90] |
Ahluwalia A. Topical glucocorticoids and the skin—mechanisms of action: an update. Mediat Inflamm, 1998, 7(3): 183-193. DOI:10.1080/09629359891126
|
| [91] |
Sun YY, Fang MR, Davies H, et al. Mifepristone: a potential clinical agent based on its anti-progesterone and anti-glucocorticoid properties. Gynecol Endocrinol, 2014, 30(3): 169-173. DOI:10.3109/09513590.2013.856410
|
| [92] |
Hanson JR. Steroids: reactions and partial synthesis. Nat Prod Rep, 2004, 21(3): 386-394. DOI:10.1039/b300379p
|
| [93] |
王玉, 张凤禹, 陈曦, 等. Thanatephorus cucumeris对可托多松19位甲基羟化反应条件的优化. 应用与环境生物学报, 2016, 22(5): 860-864. Wang Y, Zhang FY, Chen X, et al. Optimization of the transformation conditions for the 19-hydroxylation of cortexolone by Thanatephorus cucumeris. Chin J Appl Environ Biol, 2016, 22(5): 860-864 (in Chinese). |
| [94] |
Lu W, Chen X, Feng JH, et al. A fungal P450 enzyme from Thanatephorus cucumeris with steroid hydroxylation capabilities. Appl Environ Microbiol, 2018, 84(13): e00503-18.
|
| [95] |
Horinouchi M, Koshino H, Malon M, et al. Steroid degradation in Comamonas testosteroni TA441: identification of the entire β-oxidation cycle of the cleaved B ring. Appl Environ Microbiol, 2019, 85(20): e01204-19.
|
| [96] |
Zhang DL, Zhang R, Zhang J, et al. Engineering a hydroxysteroid dehydrogenase to improve its soluble expression for the asymmetric reduction of cortisone to 11β-hydrocortisone. Appl Microbiol Biotechnol, 2014, 98(21): 8879-8886. DOI:10.1007/s00253-014-5967-1
|
| [97] |
Liu YY, Wang Y, Chen X, et al. Regio- and stereoselective reduction of 17-oxosteroids to 17β-hydroxysteroids by a yeast strain Zygowilliopsis sp. WY7905. Steroids, 2017, 118: 17-24. DOI:10.1016/j.steroids.2016.11.002
|
| [98] |
Zhou YY, Wang Y, Chen X, et al. Modulating the active site lid of an alcohol dehydrogenase from Ralstonia sp. enabled efficient stereospecific synthesis of 17β-hydroxysteroids. Enzyme Microb Technol, 2021, 149: 109837. DOI:10.1016/j.enzmictec.2021.109837
|
| [99] |
Jesus M, Martins APJ, Gallardo E, et al. Diosgenin: recent highlights on pharmacology and analytical methodology. J Anal Methods Chem, 2016, 2016, 4156293.
|
| [100] |
Peng YE, Yang ZH, Wang YX, et al. Pathways for the steroidal saponins conversion to diosgenin during acid hydrolysis of Dioscorea zingiberensis C. H. Wright. Chem Eng Res Des, 2011, 89(12): 2620-2625. DOI:10.1016/j.cherd.2011.06.008
|
| [101] |
赵岩, 肖培根. 我国薯蓣属甾体激素原料植物的种质资源. 作物品种资源, 1989(1): 23-24. Zhao Y, Xiao PG. Germplasm resources of steroid hormone raw materials of dioscorea in China. China Seed Ind, 1989(1): 23-24 (in Chinese). |
| [102] |
Xu LP, Wang D, Chen J, et al. Metabolic engineering of Saccharomyces cerevisiae for Gram-scale diosgenin production. Metab Eng, 2022, 70: 115-128. DOI:10.1016/j.ymben.2022.01.013
|
| [103] |
马延和, 王钦宏, 陈五九, 等. 生产3-脱氢莽草酸大肠杆菌重组菌株及其构建方法和应用: 中国, 201711002831.2, 2017-10-24. Ma YH, Wang QH, Chen WJ. et al. Recombinant strain for producing 3-dehydrogenated shikimic acid coli as well as construction method and application thereof: CN, 201711002831.2, 2017-10-24. |
| [104] |
Tu R, Li LP, Yuan HL, et al. Biosensor-enabled droplet microfluidic system for the rapid screening of 3-dehydroshikimic acid produced in Escherichia coli. J Ind Microbiol Biotechnol, 2020, 47(12): 1155-1160. DOI:10.1007/s10295-020-02316-1
|
| [105] |
Li LP, Tu R, Song GT, et al. Development of a synthetic 3-dehydroshikimate biosensor in Escherichia coli for metabolite monitoring and genetic screening. ACS Synth Biol, 2019, 8(2): 297-306. DOI:10.1021/acssynbio.8b00317
|
| [106] |
王钦宏, 彭彦峰, 陈五九, 等. 一种生产原儿茶酸的方法: 中国, 202111135396.7. 2021-11-30. Wang QH, Peng YF, Chen WJ, et al. Method for producing protocatechuic acid: CN, 202111135396.7. 2021-11-30. |
| [107] |
王钦宏, 陈五九, 江小龙, 等. 一株生产原儿茶酸的大肠杆菌基因工程菌及其构建方法与应用: 中国, 201711390790.9, 2017-12-21(in Chinese). Wang QH, Chen WJ, Jiang XL, et al. Escherichia coli genetically-engineered bacterium for producing protocatechuic acid and construction method and application of Escherichia coli genetically-engineered bacterium: CN, 201711390790.9, 2017-12-21(in Chinese). |
| [108] |
王钦宏, 曹鹏, 陈五九, 等. 发酵法生产酪氨酸的大肠杆菌及其构建方法与应用: 中国, 201711343436.0, 2017-12-15. Wang QH, Cao P, Chen WJ, et al. The invention discloses Escherichia coli for producing tyrosine by a fermentation method as well as a construction method and an application of the Escherichia coli: CN, 201711343436.0, 2017-12-15(in Chinese). |
| [109] |
马延和, 王钦宏, 陈五九, 等. 生产左旋多巴大肠杆菌重组菌株及其构建方法与应用: 中国, 201711003046.9, 2017-10-24. Ma YH, Wang QH. Chen WJ, et al. Recombinant strain for producing levodopa coli as well as construction method and application thereof: CN, 201711003046.9, 2017-10-24(in Chinese). |