1. School of Biology and Biological Engineering, South China University of Technology, Guangzhou 510006, Guangdong, China;
2. Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, China;
3. College of Life Sciences, University of Chinese Academy of Sciences, Beijing 100049, China;
4. School of Life Sciences, Hebei University, Baoding 071002, Hebei, China
Received: February 1, 2021; Accepted: March 28, 2021; Published: December 27, 2021
Supported by: National Key Research and Development Program of China (2018YFA0902900); National Natural Science Foundation of China (31970063); Key Project of Chinese Academy of Sciences (QYZDB-SSW-SMC012); International Partnership Program of Chinese Academy of Sciences (153D31KYSB20170121); Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (TSBICIP-PTJS-003)
#There authors contributed equally to this study.
新型的碱基编辑技术结合了CRISPR/Cas系统的靶向特异性与碱基脱氨酶的催化活性,可以在引导RNA (guide RNA, gRNA) 的指引下,使用切割功能受损的Cas突变体(dCas或nCas) 与碱基脱氨酶的融合蛋白,在特定位点实现碱基的替换[1-2]。2016年,David Liu研究团队与Akihiko Kondo研究团队率先开发BE[3]和Target-AID[4]胞嘧啶碱基编辑器,实现了靶点C到T的碱基转换(transversion)。近年来,该技术不断得到完善和补充,目前已可实现A到G的碱基转换,大肠杆菌内C到A的碱基颠换(transition) 和哺乳动物细胞内C到G的碱基颠换[5-7]。该技术不产生双链切割、不需要外源模板且不依赖染色体DNA的同源重组,成为真核及原核生物中基因组编辑工具的强大补充。
谷氨酸棒杆菌是一株生物安全且具有重要工业应用的底盘微生物,目前被广泛地应用于生产多种大宗化学品[8]。在之前的研究工作中,本研究团队已在谷氨酸棒杆菌中开发了一种多元自动化的碱基编辑方法MACBETH (multiplex automated Corynebacterium glutamicum base editing method)[9],实现了靶点C到T的转换。随后,通过引入不同Cas9突变体、截短或延长靶标序列长度、引入腺苷脱氨酶,实现了对碱基编辑工具靶标范围、编辑窗口和碱基转化种类的拓展[10]。同时,通过构建基于绿色荧光蛋白(green fluorescent protein, GFP) 蛋白的检测系统,并结合流式细胞仪分析技术,对碱基编辑的培养条件、诱导条件和编辑时间等因素进行了优化,进一步提升了碱基编辑效率[11]。此外,黄华媚等在谷氨酸棒杆菌中开发了BE结构的胞嘧啶碱基编辑器[12],Deng等则在谷氨酸棒杆菌中结合胞嘧啶与腺嘌呤碱基编辑技术,开发了双功能碱基编辑器[13]。
在谷氨酸棒杆菌中,相较于基于双链断裂的CRISPR/Cas9基因编辑方法,本研究团队开发的碱基编辑技术,融合只有一条链切割活性的nCas9(D10A) 蛋白与胞嘧啶脱氨酶,对细胞产生毒性较小,不依赖同源重组,利用该优势,首次实现了谷氨酸棒杆菌3个靶基因的同时编辑,对基因组多位点改造提供了便利[9]。目前该多位点编辑方法基于多个单引导RNA (single guide RNA, sgRNA) 的转录,每个sgRNA的转录单元需要单独的启动子与终止子,常规基于同源重组的质粒构建上仍然存在较大困难。例如,如果均采用相同的启动子与终止子,会造成重复序列,质粒连接困难;如果采用不同的启动子与终止子,则会造成每个sgRNA转录水平不一致;另外,该构建方法不具有通用性,更换靶点困难。基于上述问题,本研究尝试多种策略,对谷氨酸棒杆菌中多位点碱基编辑系统进行优化:首先优化基于单独启动子/终止子多重sgRNA表达框的构建方法,通过构建框架质粒,并结合Golden Gate连接方法,避免重复序列干扰,加速靶点更换;另外,构建基于Type Ⅱ CRISPR crRNA (CRISPR RNA) 阵列、tRNA加工的多gRNA表达框,这两种形式均只需要一个启动子和一个终止子序列,简化多重gRNA表达框结构。上述研究的开展不仅为谷氨酸棒杆菌多位点编辑调控奠定了方法基础,而且也为其他原核菌株基因组改造提供了参考。
1 材料和方法
1.1 材料
1.1.1 菌种和质粒 本研究所用菌株与质粒为所在实验室购买和保存,详见表 1。
表 1 本文所用质粒与菌株
Table 1 Plasmids and strains used in this study
| Names |
Sources |
| Plasmids |
|
| pTrcmob-gRNA-ccdB |
Lab stock |
| pXMJ19-nCas9(D10A)-AID-gRNA-ccdB |
Lab stock |
| pTrcmob-upp-rfp-gRNA |
Lab stock |
| pTrcmob-upp-rfp-ald-gRNA |
Lab stock |
| pUC57-TR |
This study (gene synthesis) |
| pET28a-CR |
This study (gene synthesis) |
| pUC57-tracrRNA |
This study |
| pTrcmob-gRNA-ccdB-Multis |
This study |
| pET28a-CR-ccdB |
This study |
| pXMJ19-nCas(D10A)-AID-crRNA-ccdB |
This study |
| pXMJ19-nCas(D10A)-AID-crRNA-ccdB-tracrRNA |
This study |
| pXMJ19-upp-rfp-gRNA(array) |
This study |
| pXMJ19-upp-rfp-ald-gRNA(array) |
This study |
| pTrcmob-tRNA-gRNA-ccdB-Multis |
This study |
| pTrcmob-upp-rfp-gRNA-tRNA |
This study |
| pTrcmob-upp-rfp-ald-gRNA-tRNA |
This study |
| pTrcmob-upp-rfp-ald-citB-gRNA |
This study |
| pTrcmob-upp-rfp-ald-Cgl0121-gRNA |
This study |
| Strains |
|
| Escherichia coli DH5α |
Lab stock |
| E. coli DB3.1 |
Lab stock |
| C. glutamicum ATCC 13032 |
Lab stock |
| C. glutamicum ATCC 13032-rfp |
Lab stock |
1.1.2 酶、引物及相关试剂盒 Q5 High-Fidelity DNA聚合酶、T4 DNA连接酶、T4 Polynucleotide Kinase购自NEB公司,2×Taq Master Mix (Dye) 购自康为世纪有限公司,大肠杆菌感受态细胞制备试剂盒购自TaKaRa (大连);各种限制性内切酶购自Thermo Fisher公司;多片段同源重组试剂盒购自南京诺维赞生物科技股份有限公司(南京);PCR引物(表 2) 由擎科生物(北京) 有限公司合成;基因合成由金斯瑞(南京) 生物科技有限公司完成;质粒小量抽提试剂盒、琼脂糖凝胶DNA回收试剂盒购自天根生化科技(北京) 有限公司。
表 2 本文使用的引物
Table 2 Primers used in this study
| Primer names |
Sequences (5′→3′) |
Size (bp) |
| Pgmultis1F |
AACAGGTACAGTGTAATTCAGGTCTCAGTTTTAGAGCTAGAAATA |
45 |
| Pgmultis1R |
CACGATAAGCTGCACAAATACCTGA |
25 |
| Pgmultis2F |
TCAGGTATTTGTGCAGCTTATCGTG |
25 |
| Pgmultis2R |
GCTATTTCTAGCTCTAAAACTTATATTCCCCAGAACATCAGGTTAATG |
48 |
| Pgmultis3F |
GTTTTAGAGCTAGAAATAGCAAGTT |
25 |
| Pgmultis3R |
TGAATTACACTGTACCTGTTGCG |
23 |
| WYgold-urF |
CCAGGTCTCTTTCAGCAGCCAGGTGTGGACGCATGTTTTAGAGCTAGAAATAGCAAGTT |
59 |
| WYgold-urR |
CCAGGTCTCTAAACTCCCGGAAGGTTTCAAATGGTGAATTACACTGTACCTGTTGC |
56 |
| WYgold-ura1F |
CCAGGTCTCTTTCAGCAGCCAGGTGTGGACGCATGTTTTAGAGCTAGAAATAGCAAGTT |
59 |
| WYgold-ura1R |
CCAGGTCTCTGAAGGTTTCAAATGGTGAATTACACTGTACCTGTTGCG |
48 |
| WYgold-ura2F |
CCAGGTCTCTCTTCCGGGAGTTTTAGAGCTAGAAATAGCAAGTT |
44 |
| WYgold-ura2R |
CCAGGTCTCTAAACAGCTTGATGTTCTGAACTCGTGAATTACACTGTACCTGTTGCG |
57 |
| WYgold-ura3F |
CCAGGTCTCTTCAGAACATCAAGCTGTTTTAGAGCTAGAAATAGCAAGTT |
50 |
| WYgold-ura3R |
CCAGGTCTCTAAACCGGCACGCATGGCGCGTCGATGAATTACACTGTACCTGTTGC |
56 |
| WYgold-ura4F |
CCAGGTCTCTTCAGAACATCAAGCTGTTTTAGAGCTAGAAATAGCAAGTT |
50 |
| WYgold-ura4R |
CCAGGTCTCTAAACGCTCATTTCTAATGAGTTGCTGAATTACACTGTACCTGTTGC |
56 |
| ccdB-BsaⅠ-F |
CACAAAACTGAGACCACGCGTGGATCCGG |
29 |
| ccdB-BsaⅠ-R |
TGTGCTAATGAGACCTTATATTCCCCAGA |
29 |
| TR-F |
CCTCGCGAATGCATCTAGATTTTTCTCCACATAAGCTGGCAA |
42 |
| TR-R |
CGTTTTATTTGATGCCTGGATTACGAAATCATCCTGTGGAGC |
42 |
| TR-2*rrnB-F |
TCCAGGCATCAAATAAAACGAAA |
23 |
| TR-2*rrnB-R |
CAGTCGACGGGCCCGGGATCCGATTCGAAGCCGCACGTCATCTAGC |
46 |
| crRNA-F |
TATTTCTTAATAACTAAAAATAT |
23 |
| crRNA-R |
TCACACTACTCTTCTTTTGCCT |
22 |
| addNotⅠ-F |
TTGGGTGCACGAGCGGCCGCGTGGGTTACATCGAACTGGATC |
42 |
| p11F-cr-R |
TTTTTAGTTATTAAGAAATATGAATTACACTGTACCTGTTGC |
42 |
| cx180322-2 |
CTTGGGTGAGCTGCATGCTA |
20 |
| addNotⅠ-R |
GCGGCCGCTCGTGCACCCAACTGATCTT |
28 |
| 2*rrnB-F |
GCAAAAGAAGAGTAGTGTGATCCAGGCATCAAATAAAACGAA |
42 |
| cx180322-2R |
TAGCATGCAGCTCACCCAAG |
20 |
| NotⅠ-TR-F |
AGTTGGGTGCACGAGCGGCCTTTTCTCCACATAAGCTGGCAAT |
43 |
| NotⅠ-TR-R |
TCGATGTAACCCACGCGGCCTCGAAGCCGCACGTCATCTAGCG |
43 |
| upp-rfp-1-F |
AAACTGCGCTGGTTGCAGCCAGGTGTGGACGCATGTTTTAGAGCTATGCTGTTTTGAAT |
59 |
| upp-rfp-1-R |
GACCATTCAAAACAGCATAGCTCTAAAACATGCGTCCACACCTGGCTGCAACCAGCGCA |
59 |
| upp-rfp-2-F |
GGTCCCAAAACTGGCTCTTCACCATTTGAAACCTTCCGGGAGTT |
44 |
| upp-rfp-2-R |
CTAAAACTCCCGGAAGGTTTCAAATGGTGAAGAGCCAGTTTTGG |
44 |
| ura-2-F |
GGTCCCAAAACTGGCTCTTCACCATTTGAAACCTTCCGGGAGTTTTAGAGCTATG |
55 |
| ura-2-R |
ACAGCATAGCTCTAAAACTCCCGGAAGGTTTCAAATGGTGAAGAGCCAGTTTTGG |
55 |
| ura-3-F |
CTGTTTTGAATGGTCCCAAAACGTGGCTGAACCGAGTTCAGAACATCAAGCTGTT |
55 |
| ura-3-R |
CTAAAACAGCTTGATGTTCTGAACTCGGTTCAGCCACGTTTTGGGACCATTCAAA |
55 |
| tRNAmultis-1F |
GGTCTCAGTTTTAGAGCTAGAAATA |
25 |
| tRNAmultis-1R |
TTATATTCCCCAGAACATCAGGT |
23 |
| tRNAmultis-2F |
TGATGTTCTGGGGAATATAAGTTTTAGAGCTAGAAATAGCAAGTT |
45 |
| tRNAmultis-2R |
CTACATCCGCCGGACTAGCCTTATTTTAACTTGCT |
35 |
| tRNAmultis-3F |
GGCTAGTCCGGCGGATGTAGCGCAGTTG |
28 |
| tRNAmultis-3R |
CTAGCTCTAAAACTGAGACCTTGAGCGGATGACGAGACTC |
40 |
| T-ur-1F |
CCAGGTCTCTTTCAGCAGCCAGGTGTGGACGCATGTTTTAGAGCTAGAAATAGCAAGTT |
59 |
| T-ur-1R |
CCAGGTCTCTAAACTCCCGGAAGGTTTCAAATGGTTGAGCGGATGACGAGACTCG |
55 |
| T-ura-2F |
CCAGGTCTCTTGAAACCTTCCGGGAGTTTTAGAGCTAGAAATAGCAAGT |
49 |
| T-ura-2R |
CCAGGTCTCTAAACAGCTTGATGTTCTGAACTCGTTGAGCGGATGACGAGAC |
52 |
1.1.3 培养基 LB培养基(g/L):蛋白胨10,NaCl 10,酵母粉5。制备固体培养基时添加2%的琼脂。
BHI培养基(g/L):脑心浸液37,(NH4)2SO4 10,K2HPO4 0.2,NaH2PO4 0.3,MgSO4·7H2O 0.5,pH 7.2。
MgSO4·7H2O为50 g/L的100×储液,于使用前稀释加入。
LBHIS培养基(g/L):蛋白胨5,NaCl 10,酵母粉2.5,脑心浸液18.5,山梨醇91.1。
CGXII培养基(g/L):具体配方参见文献[11]。
NCM培养基(g/L):具体配方参见文献[14]。
0.9%的生理盐水(g/L):NaCl 9,配置成溶液,121 ℃灭菌20 min。
5-氟尿嘧啶(5-fluorouracil,5-FU):100 μmol/L,配成100×母液。
1.2 方法
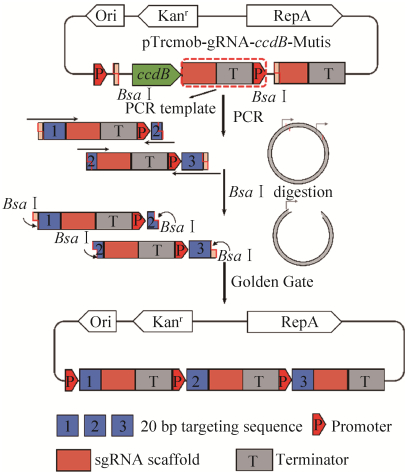
1.2.1 框架质粒构建 框架质粒pTrcmob-gRNA-ccdB-Multis的构建:以实验室保存的质粒pTrcmob-gRNA-ccdB为模板,引物Pgmultis1F与Pgmultis1R、Pgmultis2F与Pgmultis2R,分别扩增得到其中2个载体框架片段;另外,用引物Pgmultis3F与Pgmultis3R,以实验室已获得质粒pTrcmob-upp-rfp-gRNA为模板扩增得到另一个条带大小约为445 bp (含两端Bsa Ⅰ酶切位点、gRNA scaffold、rrnB终止子、P11F启动子) 的条带;将上述获得的3个片段通过同源重组连接,具体操作方法见ClonExpress® MultiS One Step Cloning Kit说明书,然后转化至E. coli DB3.1感受态;最后,经过菌落PCR以及PstⅠ、BamHⅠ和NcoⅠ进行酶切鉴定,并对质粒进行Sanger测序,获得正确质粒。
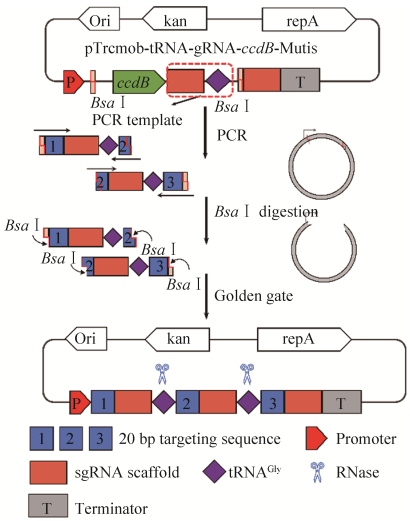
框架质粒pTrcmob-tRNA-gRNA-ccdB-Multis的构建:将质粒pTrcmob-gRNA-ccdB作为模板,用引物tRNAmultis-1F和tRNAmultis-1R扩增出大小约为5 811 bp的载体片段,用引物tRNAmultis-2F和tRNAmultis-2R扩增出第一个大小约为72 bp (含两端同源臂片段、gRNA scaffold) 的插入片段,然后以谷氨酸棒杆菌ATCC 13032基因组为模板,用引物tRNAmultis-3F和tRNAmultis-3R扩增出大小约为106 bp的第二个插入片段(含两端同源臂片段、tRNAGly),最后将载体片段与两个插入片段进行同源重组后并转化,挑单克隆培养并测序获得正确质粒pTrcmob-tRNA-gRNA-ccdB- Multis。
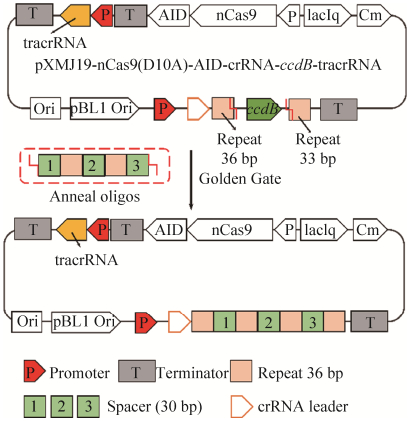
框架质粒pXMJ19-nCas9 (D10A)-AID-crRNA- ccdB-tracrRNA的构建:分别将crRNA序列、tracrRNA序列进行基因合成,序列参考文献[15]。首先,利用含合成基因的质粒pET28a- CR,通过BsaⅠ酶切获得载体片段,并利用质粒pXMJ19-nCas9-AID-gRNA-ccdB,PCR扩增得到ccdB基因,然后两片段通过Golden Gate连接后获得新质粒pET28a-CR-ccdB;然后,利用含合成基因的质粒pUC57-TR,XbaⅠ和Bam HⅠ酶切后获得pUC57载体框架,同时PCR扩增得到大小约为341 bp的tracrRNA插入片段,并且利用质粒pTrcmob-gRNA-ccdB扩增得到rrnB终止子序列,之后将3个片段同源重组后获得新质粒pUC57-tracrRNA;然后,利用上述构建的pET28a-CR-ccdB扩增出大小约为1 041 bp (crRNA片段中插入ccdB基因) 的第一个插入片段,以质粒pXMJ19-nCas9-AID-gRNA-ccdB为模板分别扩增出大小约为3 720 bp、4 501 bp、3 883 bp的载体框架片段,然后通过4个片段同源重组获得新质粒pXMJ19-nCas(D10A)-AID- crRNA-ccdB;最后,采用NotⅠ单酶切pXMJ19- nCas9(D10A)-AID-crRNA-ccdB质粒获得13 063 bp大小的载体框架片段,同时利用pUC57- tracrRNA质粒扩增出650 bp的插入片段(含P11F启动子、tracrRNA、rrnB终止子以及片段两端同源臂序列),再经过同源重组的方法即获得最终的质粒pXMJ19-nCas9(D10A)-AID-crRNA- ccdB-tracrRNA。
1.2.2 Golden Gate连接构建多基因编辑质粒 质粒pTrcmob-upp-rfp-gRNA的Golden Gate构建:以质粒pTrcmob-gRNA-ccdB-Multis为模板,以WYgold-urF和WYgold-urR为引物扩增得到片段大小约为513 bp(含两端BsaⅠ酶切位点、upp基因20 bp的靶标序列、gRNA scaffold、rrnB终止子、P11F启动子、rfp基因20 bp的靶标序列)的片段;将该片段与pTrcmob-gRNA- ccdB-Multis质粒通过Golden Gate方法进行连接,构建得到该质粒,最后经过菌落PCR以及测序验证得到该质粒。具体Golden Gate方法可参见文献[9]。质粒pTrcmob-upp-rfp-ald-gRNA、pTrcmob-upp-rfp-ald-citB-gRNA与pTrcmob-upp- rfp-ald-Cgl0121-gRNA的构建方法与上述类似。
质粒pXMJ19-upp-rfp-gRNA(array) 的构建:先将引物upp-rfp-1-F和upp-rfp-1-R,upp-rfp- 2-F和upp-rfp-2-R分别采用NEB公司的T4 Polynucleotide Kinase进行引物退火与磷酸化反应,然后将2个磷酸化退火后的片段和质粒pXMJ19-nCas9(D10A)-AID-crRNA-ccdB- tracrRNA一起进行Golden Gate,最后经过转化至E. coli DH5α,菌落PCR以及测序获得目的质粒pXMJ19-upp-rfp-gRNA (array)。质粒pXMJ19-upp- rfp-ald-gRNA (array) 的构建与上述类似。
质粒pTrcmob-upp-rfp-gRNA-tRNA的构建:以T-ur-1F和T-ur-1R为引物,以质粒pTrcmob- tRNA-gRNA-ccdB-Multis为模板扩增出约166 bp的插入片段(含两端BsaⅠ酶切位点、upp基因20 bp的靶标序列、gRNA scaffold、tRNAGly、rfp基因20 bp的靶标序列),然后将质粒pTrcmob- tRNA-gRNA-ccdB-Multis和插入片段用Golden Gate方法进行连接,通过菌落PCR以及测序得到正确的目的质粒pTrcmob-upp-rfp-gRNA-tRNA。质粒pTrcmob-upp-rfp-ald-gRNA-tRNA的构建与上述类似。
1.2.3 谷氨酸棒杆菌的转化 1) 感受态制备: 首先将−80 ℃保存的谷氨酸棒杆菌取出后,取少量菌液于固体培养基LBHIS上进行划线培养,然后再挑取适量菌转接至BHI液体培养基中,30 ℃、220 r/min过夜培养。将过夜培养的菌液以初始OD600为0.3转接到含异烟肼、Tween 80、dl-苏氨酸、甘氨酸的NCM培养基中,30 ℃摇床培养。当OD600在0.8–1时,制备感受态细胞,具体方法参见文献[14]。
2) 质粒转化:取1 μg左右的不同多位点碱基编辑质粒分别添加至80 μL感受态细胞中,混匀后转移至1 mm电击杯中,1.8 kV电转后立即加入至含1 mL BHIS培养基(46 ℃预热)的1.5 mL EP管中,并转移至金属浴锅46 ℃温浴6 min。最后,摇床30 ℃培养1–2 h后,取适量涂布于含相应抗性的LBHIS固体培养基平板上,30 ℃静置培养2–3 d,直至长出单菌落。
1.2.4 谷氨酸棒杆菌的培养及多位点碱基编辑 1) 种子培养:挑取生长良好的单菌落接种于每孔装有0.6 mL LBHIS液体培养基及相应抗生素的96孔深孔板中,30 ℃、800 r/min培养24 h。
2) 诱导培养:将种子培养液按初始OD600为0.2–0.3的接种量转接至新的每孔装有0.6 mL的LBHIS液体培养基(含相应抗生素及终浓度为1 mmol/L的IPTG) 的96孔深孔板中。然后,30 ℃高通量摇床800 r/min培养24 h。
本文所有多位点编辑实验均按上述步骤操作,没有进行后续传代。
1.2.5 编辑比例分析与克隆挑选 分别取200 μL左右上述诱导培养结束的菌液,4 000 r/min离心1 min,去除上清液后用1 mL左右0.9%的生理盐水清洗两遍,去除残留的IPTG。再用0.9%的生理盐水稀释105–106倍后涂布于相应抗性的BHI固体培养基平板上,30 ℃培养箱静置培养1–2 d,直至长出大小合适的单菌落。最后,分别将上述BHI固体培养基中的菌随机挑选24个单克隆接种于含对应抗性的LBHIS液体培养基中,800 r/min培养16 h。每种编辑系统均设置3个平行实验。
利用上述得到的单克隆菌液,分别验证多基因编辑中各个基因的失活效率。
upp基因:将各个单克隆分别在添加/不添加5-氟尿嘧啶的CGXII固体培养基上划线,upp基因失活后才能在添加5-氟尿嘧啶的CGXII固体培养基平板上生长,以此快速衡量upp基因失活效率。
rfp基因:使用酶标仪,以野生菌C. glutamicum ATCC 13032为阴性对照,C. glutamicum ATCC 13032-rfp为阳性对照,通过比较荧光值的大小即可快速验证rfp基因失活效率(激发波长为560 nm,发射波长为607 nm)。
其他基因的失活均通过菌落PCR、Sanger测序分析得到。
2 结果与分析
2.1 优化基于单独启动子/终止子的多重sgRNA表达框 基于单独启动子/终止子的多重sgRNA表达框结构烦琐,传统的构建方法需要将启动子、sgRNA scaffold、终止子等元件单独PCR,然后再通过同源重组等方法进行组装,该方法容易受重复序列影响,难以构建成功,并且更换靶点时需要再重复以上过程。针对上述问题,首先,本研究通过构建双功能的框架质粒,再结合Golden Gate方法,简化该形式的质粒构建,加速靶点的替换。
2.1.1 基于单独启动子/终止形式的框架质粒构建及Golden Gate连接 基于单独启动子/终止形式的框架质粒结构如图 1所示,一方面,质粒上包含BsaⅠ限制性内切酶识别位点及ccdB致死基因,用于Golden Gate连接及阳性克隆筛选;另一方面,质粒上包含模板序列(含sgRNA scaffold、rrnB终止子、P11F启动子),该部分用于引入不同20 bp靶标序列时的PCR扩增。在对模板序列扩增时,引物中除了包含与模板序列的结合部分,同时在5′端还依次包含BsaⅠ识别序列、要引入的20 bp靶标序列。将PCR获得的片段与框架质粒再通过Golden Gate连接,框架质粒与插入片段会相应地形成匹配的黏性末端,连接后得到目标质粒。
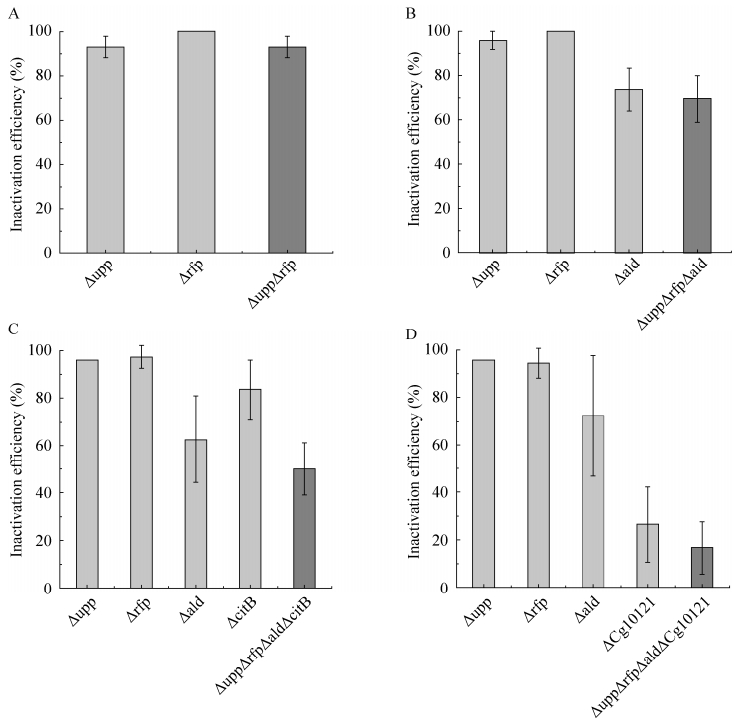
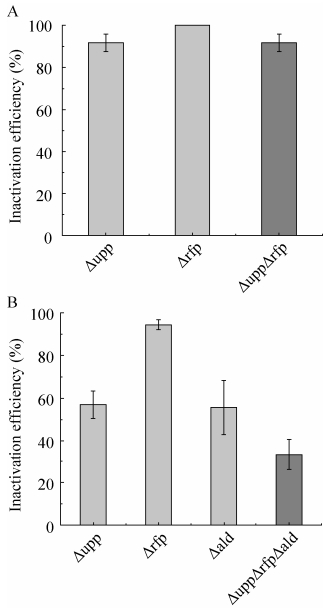
2.1.2 基因组上多基因编辑测试 利用上述框架质粒,分别构建靶向失活upp、rfp双基因的质粒,及upp、rfp、ald三基因失活的质粒(上述2个质粒与之前已报道的2个质粒序列[9]一致,只是构建方法得到优化),所用sgRNA靶标序列如表 3所示,通过表达nCas9-AID融合蛋白,使编辑框内靶标C转换为T,提前形成终止密码子,失活目的基因。相比于已报道的培养条件,本研究将培养基更换为营养更为丰富的LBHIS培养基,经过编辑后,实现了两基因和三基因的同时失活,并提高了效率,两基因失活的效率高达(93.05±4.81)% (图 2A);三基因同时失活的效率为(69.40±10.49)% (图 2B),推测效率提高较高的原因可能为丰富培养基成分更利于细胞生长、nCas9-AID蛋白的表达或编辑过程中形成单链切口的修复等。由于三基因同时失活的效率仍然较高,本研究尝试对4个基因同时编辑,分别选用citB及Cgl0121基因作为第4个靶点。补充citB后,四基因同时失活的效率为(50.00±11.02)%。而补充Cgl0121基因后,四基因同时失活的效率仅为(16.67±11.03)%,此处失活效率下降主要为Cgl0121基因靶标C位点部分被编辑为G或A,这部分编辑未被统计到失活效率中,经统计,Cgl0121基因总编辑效率为100%。由图 2C–2D可以看出,增加第4个基因后,对upp、rfp、ald三基因的失活效率影响并不大,造成上述两种情况差异较大的原因,主要为citB及Cgl0121基因单独失活效率差异较大,由此推断,不同基因的单独编辑效率对多基因编辑效率影响较大。
表 3 本文所编辑的基因及靶点
Table 3 Genes and 20 bp targeting sequence used in this study
| Genes |
20 bp targeting sequences |
PAM |
Type of early stop codons |
| upp |
GCAGCCAGGTGTGGACGCAT |
TGG |
TAG |
| rfp |
CCATTTGAAACCTTCCGGGA |
AGG |
TAA/TAG |
| ald |
CGAGTTCAGAACATCAAGCT |
GGG |
TGA |
| citB |
TCGACGCGCCATGCGTGCCG |
GGG |
TGA |
| Cgl0121 |
GCAACTCATTAGAAATGAGC |
TGG |
TAA |
| Note: the underlined Cs in 20 bp targeting sequence indicate the target editing sites. |
2.2 建立基于Type Ⅱ CRISPR crRNA阵列的多重gRNA表达框 上述优化能够有效地简化质粒的构建过程,但并没有优化多重gRNA烦琐的结构。建立基于Type Ⅱ CRISPR crRNA阵列的多重gRNA表达框能有效解决该问题。在CRISPR/Cas9系统中,crRNA阵列由一系列高度保守的约为36 bp的同向重复序列(direct repeat) 与序列特异的约为30 bp间隔序列(spacer) 串联交替组成。在crRNA成熟过程中,crRNA阵列会转录形成前体CRISPR RNA (precursor crRNA, pre-crRNA),同时转录出与pre-crRNA中重复序列互补配对的反式激活crRNA (trans-activating crRNA, tracrRNA),pre-crRNA与tracrRNA的互补配对能够触发体内核酸酶RNase Ⅲ的切割机制,产生一系列定位不同靶点的双引导RNA[16]。目前所用的sgRNA即通过融合crRNA的3′端与tracrRNA的5′端得到的序列。由于crRNA阵列中同向重复序列与间隔序列长度均较短,将该策略应用于多位点编辑时,将省去靶标序列的PCR过程,既简化了质粒构建过程,又节省了成本。目前该策略在大肠杆菌、酿酒酵母等中已有应用[17-18],在谷氨酸棒杆菌中为首次研究。
2.2.1 框架质粒构建及Golden Gate连接 首先,对已报道的crRNA阵列及tracrRNA序列进行合成,分别用组成型启动子P11F启动子及rrnB终止子表达2个元件。为方便不同间隔序列的快速插入,在crRNA阵列中插入BsaⅠ识别序列及ccdB致死基因,方便后续Golden Gate构建(图 3)。对于不同间隔序列和重复序列的插入,只需要通过合成约50 bp长度的引物,在引物两端引入相互匹配的黏性末端,通过引物磷酸化、退火结合形成双链,再与框架质粒进行Golden Gate连接,得到目标质粒。该方法省去了靶标序列的PCR过程,极大地简化了质粒的构建。对于RNase Ⅲ,则采用谷氨酸棒杆菌的内源基因(Cgl2074) 表达。
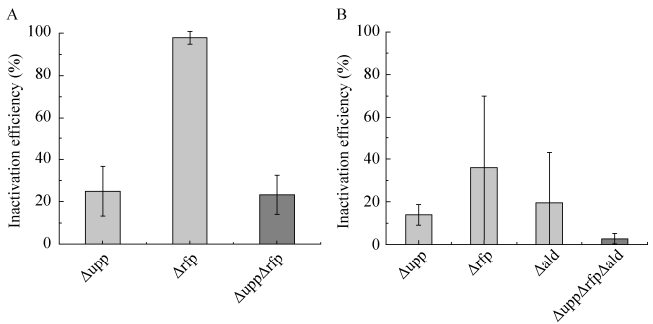
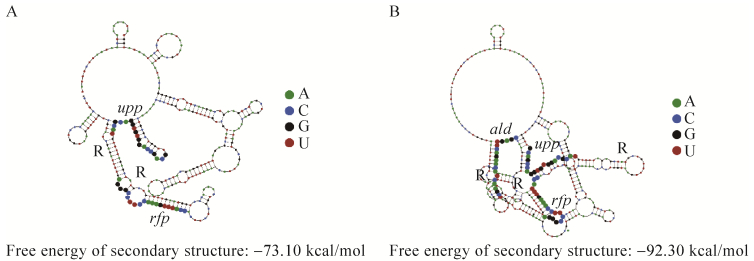
2.2.2 两基因及三基因编辑 应用上述框架质粒,分别构建靶向失活upp、rfp双基因的质粒,及upp、rfp、ald三基因失活的质粒,转化到谷氨酸棒杆菌中进行碱基编辑。如图 4所示,该策略可实现每个基因的单独失活及2个基因的同时失活,但相比于单独启动子/终止子sgRNA表达框的形式,失活效率下降明显,两基因同时失活效率仅为(23.20±9.20)%,三基因同时失活效率仅为(2.80±2.42)%。我们猜测以下两方面都可能对编辑效率产生明显影响:首先,pre-crRNA中的重复序列与tracrRNA结合形成发卡结构是crRNA成熟过程中的重要环节,最新的研究表明,如果重复序列位于pre-crRNA二级结构的复杂结构区,将影响crRNA的形成[18]。本研究对靶向两基因和三基因的pre-crRNA通过NUPACK软件(www.nupack.org) 进行二级结构预测(图 5),发现重复序列均位于复杂结构区,尤其是三基因序列,因此推测,该方面原因可能是导致编辑效率下降的原因。另外,RNase Ⅲ对pre-crRNA与tracrRNA复合体的切割也是crRNA成熟过程中的重要一步,本研究尝试过表达谷氨酸棒杆菌内源RNase Ⅲ来提高编辑效率,但并没有效果(数据未展示)。
2.3 建立基于tRNA加工的多重sgRNA表达框 建立基于Type Ⅱ CRISPR crRNA阵列的多重gRNA表达框能够有效地简化质粒构建,但编辑效率下降明显,本部分尝试建立基于tRNA加工的多重sgRNA表达框,进一步优化多重gRNA结构。该策略将菌体中内源性的tRNA加工系统与多个sgRNA结构相结合,采用一个启动子和终止子,转录形成一条RNA序列,在含不同靶点的sgRNA之间插入tRNA序列,内源性的RNase会特异性地识别tRNA序列,并进行切割,从而释放不同的sgRNA。该方法不需要引入外源蛋白,同时tRNA序列较小(约70 bp),不仅可以简化质粒结构,同时能实现质粒的快速高效构建。目前该策略已在酿酒酵母、水稻、果蝇等真核生物中应用[19-21],据笔者所知,在谷氨酸棒杆菌中为首次研究。
2.3.1 基于tRNA加工形式的框架质粒构建及Golden Gate连接 在质粒构建时,选用P11F启动子与rrnB终止子,实现RNA序列的转录,tRNA则选用常用且序列较短(76 bp) 的内源tRNAGly。该质粒不仅包含用于Golden Gate连接的元件(ccdB基因、BsaⅠ酶切位点),同时也包含PCR扩增含靶点序列时的模板序列(sgRNA scaffold、tRNAGly序列),如图 6所示。通过设计引物,在对模板序列扩增时引入20 bp的靶标序列及BsaⅠ识别序列,再与框架质粒进行Golden Gate连接,得到目标质粒。
2.3.2 两基因及三基因编辑 应用该策略,分别构建靶向失活upp、rfp双基因的质粒,及upp、rfp、ald三基因失活的质粒,然后转化至谷氨酸棒杆菌中进行碱基编辑。如图 7所示,该策略可实现两基因和三基因的同时失活,相比于单独启动子/终止子sgRNA表达框的形式,两基因的失活效率相近,高达(91.67±4.15)%,三基因同时失活的效率则有所下降,为(33.33±7.22)%。后续将深入研究谷氨酸棒杆菌中tRNAGly的成熟机制,结合cRT-PCR (circularized reverse transcription PCR)[21]及测序技术,明确tRNAGly序列的切割位点,评估单个sgRNA的释放水平,或尝试其他种类tRNA序列,进一步提高编辑效率。
3 讨论 由于谷氨酸棒杆菌具有重要的工业应用价值,针对该菌株的遗传改造技术也日新月异,从传统的自杀质粒介导的同源重组、Cre/loxP介导的位点特异性重组、RecT介导的单链重组等编辑技术,到现阶段强大的CRISPR技术,这些均在该菌株中得到开发并推动了菌株的基础与应用研究。目前,CRISPR技术在谷氨酸棒杆菌中的应用主要分为CRISPR干扰(CRISPR interference, CRISPRi)、基因敲除与插入、碱基编辑等方面[22-23]。基于双链断裂的CRISPR技术由于对菌体产生的毒性较大,在多位点编辑方面较困难,目前仅有报道通过结合CRISPR/Cas9与单链DNA重组技术,实现了双基因的同时编辑,效率为40%,但是,双基因编辑显著降低了获得的克隆数(约10个),未能获得更多位点的编辑[24]。相比于基于双链断裂的CRISPR技术,碱基编辑方法不产生双链断裂、不依赖染色体DNA同源重组、结构简单、操作方便,并且解决了传统CRISPR技术难以实现多于2个靶点同时编辑的问题。
针对多位点编辑技术,多种多重gRNA的表达策略已被开发[25],主要分为以下3类:第一类为每个sgRNA转录单元都需要单独启动子/终止子,目前绝大多数微生物中多位点碱基编辑方法均采用该形式。Banno等应用该形式已实现大肠杆菌内6个基因同时编辑(碱基编辑),效率高达87.5%[26];Zhong等在阿维链霉菌Streptomyces avermitilis中实现5个基因(效率为60%)和9个基因(效率未显示) 的同时编辑(碱基编辑)[27];Yu等则在枯草芽孢杆菌(Bacillus subtilis) 中实现3–4个基因同时编辑(碱基编辑),效率分别为100%和50%[28]。由此可见,该方法在多位点编辑中效率较高,应用较广,但结构较大,存在重复序列,质粒不稳定;第二类基于原始CRISPR/Cas系统中的crRNA阵列,通过一个启动子和一个终止子,实现对多个间隔序列及重复序列的转录,其中Cas12a及Cas13a可以自行加工crRNA阵列,释放单个成熟的gRNA,而Cas9则需要tracrRNA及RNase Ⅲ的参与。Li等在谷氨酸棒杆菌中应用CRISPR/dCas12a及crRNA阵列实现对4个赖氨酸合成途径基因的同时抑制(CRISPRi),4个基因抑制效率均高于90%[29];Bao等在酿酒酵母中应用CRISPR/Cas9及crRNA阵列实现对3个基因的同时编辑(disruption),效率高达100%[30]。而crRNA阵列在多位点碱基编辑方面,尚未有该方面的报道。该方法结构简单,方便快速构建,但在本文的应用中,效率较低,后续需要进一步优化;第三类为多个sgRNA结构由一个启动子和一个终止子转录,在每个sgRNA结构之间加入RNA切割位点,该切割方式可以基于内源的核糖酶、tRNA加工及外源的Csy4蛋白。Tong等在天蓝色链霉菌Streptomyces coelicolor中应用CRISPR-BEST及基于Csy4蛋白的多重sgRNA,实现了3个基因的同时编辑(碱基编辑),效率为33.3%[31];Zhang等在酿酒酵母中应用CRISPR/ Cas9及基于tRNA加工系统的多重sgRNA,实现了对8个基因的同时编辑(disruption),效率高达87%[21]。与其他两种方法相比,本研究选用的tRNA加工系统具有不需要引入可能对菌体有毒的外源蛋白、序列较短(约70 bp)、在多位点编辑中更稳定的优势[32]。并且,在真核生物的应用中,tRNA序列可被内源性的RNase P及RNase Z特异性地识别,切割位点较明确,释放的单个sgRNA只在3′端有1–4个碱基的残留,对sgRNA结构影响小,tRNA也可以增加Pol Ⅱ/Pol Ⅲ启动子的转录效率[20]。而在原核生物中,tRNA成熟机制研究相对较少,涉及的内源性的酶等略有不同[33]。后续将深化研究谷氨酸棒杆菌中的tRNA成熟及切割机制,为进一步提高效率提供参考。
本研究对谷氨酸棒杆菌中多位点碱基编辑系统进行了优化,对不同的多重gRNA表达策略进行构建并比较(表 4)。首先,优化基于单独启动子/终止子的多重sgRNA表达框,通过构建框架质粒,并结合Golden Gate连接方法,避免了重复序列的干扰,加速质粒构建及靶点更换,该形式编辑效率最高,但并没有简化多重sgRNA的烦琐结构;其次,建立基于Type Ⅱ CRISPR crRNA阵列的多重gRNA表达框,该策略能有效简化多重gRNA表达框,简化质粒的构建过程,降低成本,但两基因及三基因同时编辑效率下降明显;最后,建立基于tRNA加工的多重sgRNA表达框,该策略简化了多重sgRNA的结构,方便质粒构建,并且,相比基于单独启动子/终止子的多重sgRNA表达形式,该策略两基因编辑效率相差不大,三基因编辑效率有所下降,后续将深化研究谷氨酸棒杆菌中tRNA加工过程,评估释放的单个sgRNA水平,进一步对该方法进行优化。基于不同策略构建工作量及多位点编辑效率的综合考虑,谷氨酸棒杆菌中两位点同时编辑可以优先选择基于tRNA加工的多重sgRNA表达框,而更多位点的编辑可以优先选择优化后的基于单独启动子/终止子的多重sgRNA表达框构建形式。上述建议主要基于本文的研究结果,由于不同位点编辑效率的差异以及技术的不断革新,实验者需要依据自己的实验目的进行选择。综上所述,本研究对谷氨酸棒杆菌中基于CRISPR/Cas9的多位点碱基编辑系统的优化,丰富了该菌株的遗传改造方法,为代谢工程和合成生物学研究提供技术支持。
表 4 本文所用多重gRNA表达策略的比较
Table 4 Comparison of multiple gRNA expression cassettes based on different methods
Methods of
gRNA processing |
Targeting genes |
Inactivation efficiency (one passage) |
Construction methods/efficiency |
sgRNA cassettes with individual
promoters/terminators
(previous study[9]) |
upp, rfp |
(41.70±6.90)% |
Homologous recombination/low |
| upp, rfp, ald |
16.67% |
Homologous recombination/low |
| sgRNA cassettes with individual promoters/terminators (this study) |
upp, rfp |
(93.05±4.81)% |
Golden Gate/high |
| upp, rfp, ald |
(69.40±10.49)% |
Golden Gate/high |
| upp, rfp, ald, citB |
(50.00±11.02)% |
Golden Gate/high |
| upp, rfp, ald, Cgl0121 |
(16.67±11.03)% |
Golden Gate/high |
| gRNA cassettes based on Type Ⅱ CRISPR crRNA arrays |
upp, rfp |
(23.20±9.20)% |
Golden Gate/high |
| upp, rfp, ald |
(2.80±2.42)% |
Golden Gate/high |
| sgRNA cassettes based on tRNA processing |
upp, rfp |
(91.67±4.15)% |
Golden Gate/high |
| upp, rfp, ald |
(33.33±7.22)% |
Golden Gate/high |
 2022, Vol. 38
2022, Vol. 38