Efficient biosynthesis of γ-aminobutyric acid by rationally engineering the catalytic pH range of a glutamate decarboxylase from Lactobacillus plantarum
γ-氨基丁酸(γ-aminobutyric acid, GABA)是一种四碳的非蛋白质氨基酸[1],普遍存在于植物、动物和微生物中[2]。因其可以保护植物免受各种病原体侵害[3]、参与人体内血压、心率等心血管疾病的调节[4],还能作为工业生产尼龙合成前体的原料[5]而被广泛应用于食品添加剂、畜牧[6]、医药和化工等领域[7]。尽管GABA生产方法多样,包括植物富集、化学合成和生物合成法。但考虑到大批量工业化生产及绿色合成需求,生物合成法生产GABA具有广阔的应用前景[8]。生物合成法又分为直接发酵法和酶(或细胞)催化法。2015年,Pham等[9]通过引入合成蛋白支架、敲除突变和失活竞争性代谢网络构建大肠杆菌(Escherichia coli) XL1工程菌株,以10 g/L葡萄糖发酵获得GABA浓度为1.3 g/L。2022年,Zhang等[10]通过模型引导的代谢工程策略改造谷氨酸棒杆菌(Corynebacterium glutamicum) ATCC 13032,工程菌株补料分批发酵产GABA达23.07 g/L。目前GABA发酵法生产产量还无法和酶催化法相媲美,且发酵法通过微生物代谢直接发酵原料,存在下游分离提取过程复杂、生产周期长等问题。因此全细胞转化一步法合成GABA受到国内外研究者的广泛重视[11]。
谷氨酸脱羧酶(glutamate decarboxylase, GAD, EC: 4.1.1.15)是一种独特的Ⅱ类氨基酸脱羧酶,广泛分布在真核生物和原核生物中。因其在许多微生物中的抗酸性[12],对生物体维持正常的生理值具有重要作用。GAD作为生物合成法制备GABA的限速酶,在5-磷酸吡哆醛(pyridoxal-5-phosphate, PLP)少量添加下,专一性地催化l-谷氨酸脱羧生成GABA和CO2,反应过程不可逆[13]。然而,自然界中天然GAD生产者大多本底表达量不高,培养条件较难控制等使得GABA产量和生产效率较低。2013年,Jiang等[14]从猕猴桃表面分离出一株产GABA能力较强的酿酒酵母(Saccharomyces cerevisiae) MJ2,28 ℃培养约48 h,GABA产量仅有5.823 g/L。乳酸菌作为GABA主要生产菌株[15],但其天然GAD活力较低。2010年,Kook等[16]为了提高乳酸菌催化合成GABA的能力,在一株清酒乳杆菌(Lactobacillus sakei) B2-16中过表达植物乳杆菌(Lactobacillus plantarum) ATCC 14917来源的gad基因,提高了GABA的产量,重组菌产GABA最大浓度达265.3 mmol/L。但仅通过表达基因提高蛋白表达水平仍是不够的,目前利用基因工程与蛋白工程技术的手段,从分子水平改进GAD酶,使其具有优良工业属性的相关研究正在大量展开。一方面是针对不同来源GAD酶催化效率低、热稳定性差不适合持续化生产进行的相关研究,如2016年,Liu等[17]通过定点诱变巨大芽孢杆菌(Bacillus megaterium)来源的新型谷氨酸脱羧酶BmGAD提高了酶的催化活性及Vmax值。2020年,Zhang等[18]通过序列分析和自由能计算结果,突变短乳杆菌(Lactobacillus brevis) CGMCC No.1306来源的GAD酶热稳定性关键位点,获得了具有较高热稳定性且活性增加1.67倍的突变体S325A。2020年,汪钟[19]通过分析L. brevis CGMCC 1306来源的GAD酶中loop区域的β-转角结构类型,对酶进行分子改造,提高了酶的热稳定性。另一方面则是针对GAD酶作用pH偏酸性的特点,改造其pH特性的研究。如2013年,Kang等[20]通过增减E. coli来源的GAD酶结构C末端长度,获得突变体GADΔ466,大幅提升了酶的pH作用范围。2014年,Shi等[21]结合定向进化和理性突变手段改造了L. brevis Lb85来源的GAD酶,获得组合突变体T17I/D294G/E312S/ Q346H,提高了酶在pH 6.0条件下的催化活性,进一步在C. glutamicum ATCC 13032中表达突变体成功提高了GABA生产量。同年,Shin等[22]依据L. plantarum ATCC 14917来源的GAD酶结构模型,截取酶C端残基后使酶在pH 4.0–8.0的广泛范围内表现出明显的催化活性。通过提高酶在偏中性环境中的催化活力,不仅能减轻酸环境对菌体量积累的胁迫作用,还能减少环境维持剂如酸碱、无机盐的投加,从而更好地满足中性环境作业需求,降低生产成本[23]。
尝试对不同来源的GAD酶实施pH改造并构建用于GABA合成的单细胞工程菌株是具有重要生产意义的。本研究首先将一株早期经实验室常压室温等离子体(atmospheric room temperature plasma, ARTP)诱变的植物乳杆菌来源的谷氨酸脱羧酶LpGAD酶基于蛋白表面电荷理性突变后,拓宽了酶的pH适用范围并提高了酶的催化活性。由于谷氨酸棒杆菌被认为是食品安全菌株(generally regarded as safe, GRAS),因此选择谷氨酸棒杆菌作为底盘细胞,通过在胞内过表达突变体,摇瓶转化实验优化反应条件后进行5 L发酵罐全细胞转化实验,实现了l-谷氨酸到GABA的高效合成。本研究旨在解决菌体催化活性随转化液pH的升高而降低致使生产持续性及底物转化率偏低的问题,提高菌体产GABA能力,构建一种经济高效的生物催化方法,为其工业化生产奠定基础。
1 材料与方法
1.1 材料
1.1.1 菌株和质粒 大肠杆菌E. coli BL21(DE3)、植物乳杆菌HG-06[24]和谷氨酸棒杆菌C. glutamicum E01[25]由本实验室保藏。pET-28a表达载体(质粒抗性:KanR)和pXMJ19表达载体(质粒抗性:ChlR)由本实验室保存。
1.1.2 酶和试剂 限制性内切酶、DNA聚合酶、DNA Marker购自TaKaRa公司;蛋白Marker购于上海碧云天生物技术有限公司;质粒提取试剂盒、PCR产物纯化试剂盒、同源重组试剂盒和蛋白浓度测定试剂盒购自上海GENEray公司;抗生素(Kan和Chl)、异丙基β-d-硫代半乳糖苷(isopropyl-β-d-thiogalactoside, IPTG)购自生工生物工程(上海)股份有限公司;谷氨酸和γ-氨基丁酸(分析纯)购自上海阿拉丁试剂有限公司;其余培养基试剂均购自上海国药试剂有限公司。
1.1.3 主要设备 SBA-40D型生物传感仪,山东省科学院生物研究所;Thermo UltiMate 3000高效液相色谱仪,赛默飞世尔科技公司;Gene Pulser Xcell电穿孔仪,Bio-Rad公司;5 L发酵罐,上海迪必尔生物有限公司;UV-1200型可见紫外分光光度计,上海翱艺仪器有限公司;小型台式离心机和干式金属浴,Eppendorf公司;AKTA蛋白纯化仪,Cytiva有限公司;超声波破碎仪JY92-IIN,宁波新芝生物科技股份有限公司。
1.2 方法
1.2.1 引物设计 通过提取植物乳杆菌全基因组核酸序列,PCR扩增获得Lpgad基因,实验设计含同源臂引物参见表 1。引物由苏州Azenta公司合成。
表 1 本研究使用的引物
Table 1 Primers used in this study
| Primer names |
Primer sequences (5′→3′) |
Size (bp) |
| S24R-F/P2 |
GTCTTTGGTGCGCCTAGAGAACAACATGATCTT |
33 |
| S24R-R/P3 |
AAGATCATGTTGTTCTCTAGGCGCACCAAAGAC |
33 |
| D88R-F/P4 |
CGAAAGAATGCCATCAGAAAATCTGAGTACCCC |
33 |
| D88R-R/P5 |
GGGGTACTCAGATTTTCTGATGGCATTCTTTCG |
33 |
| L135K-F/P6 |
TGTATGTTAGGCGGTAAAGCAATGAAATTCGCC |
33 |
| L135K-R/P7 |
GGCGAATTTCATTGCTTTACCGCCTAACATACA |
33 |
| E170R-F/P8 |
TATCAAGTTTGCTGGAGAAAGTTTTGTGTCTAC |
33 |
| E170R-R/P9 |
GTAGACACAAAACTTTCTCCAGCAAACTTGATA |
33 |
| H196R-F/P10 |
GTCCTTGACGTTAACAGAGTCTTAGACTACGTG |
33 |
| H196R-R/P11 |
CACGTAGTCTAAGACTCTGTTAACGTCAAGGAC |
33 |
| A225K-F/P12 |
ATATGACGACCTAGCCAAACTCGATAAGGTCGTT |
34 |
| A225K-R/P13 |
AACGACCTTATCGAGTTTGGCTAGGTCGTCATAT |
34 |
| Y309K-F/P14 |
AGTCTTCAAAGTTAGTAAATTAGGTGGGGAGTTG |
34 |
| Y309K-R/P15 |
CAACTCCCCACCTAATTTACTAACTTTGAAGACT |
34 |
| A359R-F/P16 |
GCCCGCTACCTGGCAAGAGCTCTGGATAAAGTT |
33 |
| A359R-R/P17 |
AACTTTATCCAGAGCTCTTGCCAGGTAGCGGGC |
33 |
| E417K-F/P18 |
ATCCTTTCCCTGCTAATCTGAAACAACAAGTCATCCAA |
38 |
| E417K-R/P19 |
TTGGATGACTTGTTGTTTCAGATTAGCAGGGAAAGGAT |
38 |
| LpGAD-F/P1 |
ATGGGTCGCGGATCCGAATTCATGGCAATGTTATACGG |
38 |
| LpGAD-R/P20 |
TCGAGTGCGGCCGCAAGCTTTCAGTGTGTGAATAGGTATT |
40 |
| pXMJ19-Lpgad-F |
AGAATTAATTAAGCTTAAAGGAGGGAAATCATGGCAATGTTATAC |
45 |
| pXMJ19-Lpgad-R |
CAAAACAGCCAAGCTGAATTCTCAGTGTGTGAATAGGT |
38 |
| The italic sequences are the mutation sites and the restriction enzyme cutting sites. |
1.2.2 培养基与培养方法 LB培养基(g/L):蛋白胨10,氯化钠10,酵母粉5。121 ℃灭菌20 min,用于大肠杆菌培养。
脑心浸液(brain-heart infusion, BHI)培养基(g/L):脑心浸液粉35。115 ℃灭菌20 min,用于谷氨酸棒杆菌培养。
MRS培养基(g/L):蛋白胨10,牛肉膏8,葡萄糖20,酵母粉4,乙酸钠5,柠檬酸氢二胺2,吐温-80 1 mL,磷酸氢二钾2,硫酸镁0.2,一水硫酸锰0.05。115 ℃灭菌20 min,用于植物乳杆菌培养。
1.2.3 重组菌株的构建 大肠杆菌重组菌构建:以植物乳杆菌基因组为模板,通过PCR扩增获得谷氨酸脱羧酶基因片段Lpgad,与经限制性内切酶(EcoR Ⅰ/Hind Ⅲ)双酶切后的pET-28a质粒片段进行同源重组酶连,连接产物转化至E. coli BL21(DE3)感受态细胞,转化子进行菌落PCR验证,挑取阳性转化子进行培养,提取质粒送测序验证。
大肠杆菌突变菌构建:以重组质粒pET28a-Lpgad为模板,根据突变位点上下游的引物对(表 1)分别PCR扩增得到含突变碱基的上下游基因片段,利用融合PCR获得完整突变基因片段,目的片段与双酶切后的pET-28a质粒片段同源重组酶连,转化至E. coli BL21(DE3)感受态细胞中。转化子菌落PCR验证条带大小正确后,提取质粒送测序验证。
谷氨酸棒杆菌重组菌构建:以重组质粒pET28a-Lpgad为模板,PCR扩增获得带质粒酶切位点两侧同源臂的基因片段,与经限制性内切酶(EcoR Ⅰ/Hind Ⅲ)双酶切后的质粒pXMJ19同源重组酶连,转化E. coli BL21(DE3)感受态细胞,构建E. coli BL21/pXMJ19-Lpgad菌株,提取转化子质粒送测序验证成功后,重组质粒电击转化C. glutamicum E01感受态细胞[26],构建C. glutamicum E01/pXMJ19-Lpgad菌株。
1.2.4 GAD的诱导表达与纯化 验证成功后的转化子液体扩大培养,当OD600达到0.6−0.8即大肠杆菌对数生长期时,加入终浓度1.0 mmol/L IPTG,16 ℃过夜诱导蛋白表达。4 ℃、8 000 r/min离心6 min收集菌体,50 mmol/L、pH 7.4的磷酸缓冲盐溶液(phosphate buffered saline, PBS)悬浮菌体并进行细胞破碎。细胞破碎液经4 ℃、12 000 r/min离心20 min后得到粗酶液,利用1 mL镍亲和柱进行纯化。0−500 mmol/L咪唑线性梯度洗脱后,收集目标蛋白,加入10% (体积分数)甘油,–40 ℃保存。SDS-PAGE (12%丙烯酰胺)分析蛋白表达及纯化情况。
1.2.5 酶活检测方法 GAD酶活测定方法[27]:反应体系(1 mL)含有0.2 mol/L柠檬酸-柠檬酸钠缓冲液,10 mmol/L底物l-谷氨酸和100 μmol/L PLP。加入50 μL酶液到1 mL预热的酶活测定缓冲液中启动反应。37 ℃保温10 min后,沸水浴中加热10 min终止反应,离心收集样品。GABA和底物l-谷氨酸浓度分别用液相色谱仪和SBA生物传感仪测定。谷氨酸脱羧酶酶活单位(U)定义为1 min产生1 μmol γ-氨基丁酸所需的酶量。
蛋白浓度通过考马斯亮蓝染色法(Bradford法)测定[28]。GAD酶比活力为每毫克蛋白质所具有的酶活力单位数,单位为U/mg。以测得酶最高比活力的条件作为酶最适反应条件,将纯酶放入不同pH缓冲液和不同温度保温处理前的酶活力值定义为100%,通过测定残余酶活力分析酶的pH稳定性和温度稳定性。
1.2.6 突变位点选取方法 将LpGAD蛋白序列提交至SWISS-MODEL[29]服务器(https://swissmodel.expasy.org/)建模获得PDB结构,通过PROCHECK[30]评估后(位于禁止区域的氨基酸数量小于氨基酸总数的5%)使用Rosetta Supercharge (https://www.rosettacommons.org/)进行基于表面电荷的蛋白理性设计[31],通过上调电荷10−20 kcal/mol,得到9个表面残基候选位点(表 1)。突变位点选取考虑表面相互作用,包括静电和疏水相互作用,以Lys或Arg进行替代,允许通过突变疏水氨基酸来调节蛋白的表面电位分布。
1.2.7 分子对接及动力学模拟方法 利用Schrödinger2020-3软件包中的Glide模块分子对接构建LpGAD、辅助因子PLP及谷氨酸的三元复合物,进一步通过计算机点突变的方式获取突变体的三元复合物。模拟全程使用Amber18软件包[32]完成。辅助因子PLP的力场参数采用antechamber计算,其中电荷信息采用键电荷校正。蛋白体系采用ff14SB力场,用Sander模块对体系进行能量最小化。随后依次进行500 ps的正则系宗模拟,1 ns的等温等压系宗(constant-pressure, constant-temperature, NPT)模拟和轨迹收集阶段时50 ns的NPT模拟。非键截止距离设定为8 Å,用particle mesh Ewald方法计算长程静电作用[33],涉及氢原子键的键长采用Shake设置进行限制[34],温度控制使用Langevin算法[35],其中碰撞频率γ设为2/ps。体系压强为1 atm,积分步长为2 fs,每隔10 ps保存轨迹,输出的轨迹用于进一步分析计算。
1.2.8 全细胞转化条件 摇瓶转化条件:细胞培养结束后收集菌体,重悬于0.9% (质量体积分数) NaCl溶液中,控制菌体量OD600=20,l-谷氨酸浓度100 g/L,辅助因子PLP 100 μmol/L,终体积50 mL转化体系不控pH,在37 ℃、200 r/min条件下转化20 min后取样。
5 L发酵罐转化条件:细胞培养结束后收集菌体,重悬于0.9% (质量体积分数) NaCl溶液中,控制菌体量OD600为20,在0 h投入l-谷氨酸100 g,辅助因子PLP 100 μmol/L,终体积1 L转化体系不控pH,在40 ℃、300 r/min条件下进行转化。反应过程中及反应结束后取样,分析产物及底物含量。
1.2.9 HPLC检测条件 产物GABA的HPLC测定方法:取反应液用5%三氯乙酸(trichloroacetic acid, TCA)稀释5倍,在4 ℃冰箱沉淀蛋白3 h左右离心,取上清液用0.22 μm膜过滤处理收集样品后,采用OPA在线衍生法分析样品。色谱柱:diamonsil C18 (5 μL, 250 mm×4.6 mm);流动相A:醋酸钠8 g/L、四氢呋喃5 mL/L、三乙胺0.225 mL/L,pH 7.2;流动相B:醋酸钠6 g/L、甲醇400 mL/L、乙腈400 mL/L,pH 7.2;检测器:UV Detector;检测波长:338 nm;柱温:40 ℃;进样量:10 μL;流速:1.0 mL/min。梯度洗脱过程:0–22.5 min,8% B;22.5−22.6 min,8% B→60% B;22.6−26.5 min,60% B;26.5− 31.5 min,100% B;31.5−35.0 min,8% B。
2 结果与分析
2.1 谷氨酸脱羧酶性质研究及分子改造
2.1.1 谷氨酸脱羧酶基因的克隆与表达 GAD是催化GABA生产的关键酶,以实验室一株ARTP诱变后的植物乳杆菌GB HG-06基因组[24]全序列为模板,PCR扩增获得Lpgad基因序列,长度为1 410 bp (图 1A),按照1.2.3方法构建重组菌E. coli BL21/pET28a-Lpgad并进行诱导表达,SDS-PAGE分析结果显示,在52 kDa有明显条带加粗,这表明LpGAD在E. coli BL21中成功实现了表达(图 1B)。
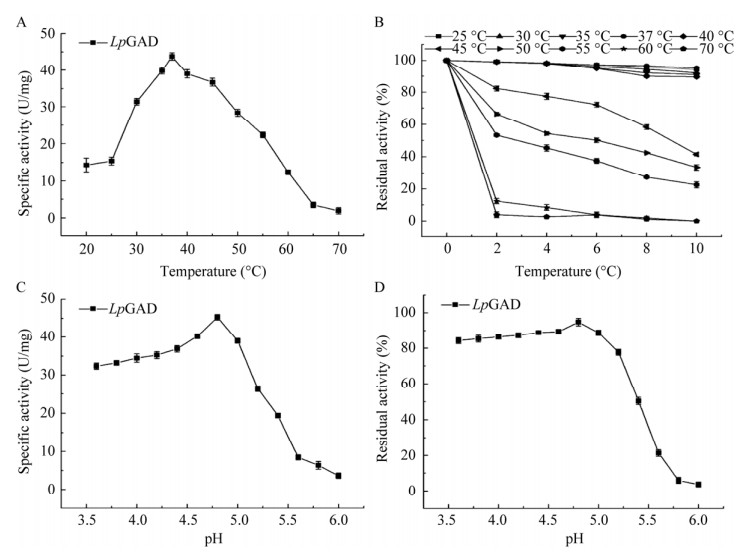
2.1.2 谷氨酸脱羧酶的酶学性质研究 不同温度条件下谷氨酸脱羧酶LpGAD的酶活测定结果显示,LpGAD的最适温度为37 ℃,高于55 ℃时酶活力急剧下降,30−45 ℃范围内能维持70%以上的酶活力(图 2A);40 ℃以下保温10 h后,残余酶活保持在85%以上,具有良好的热稳定性(图 2B)。pH测定结果显示,最适pH为4.8,酶在pH 3.6−5.0下的活力均保持在60%以上,当pH高于5.0时酶活力急剧下降,pH 6.0以上几乎检测不到酶活(图 2C),这表明LpGAD最佳催化pH偏酸性,偏中性范围内催化能力极差甚至没有,这与以往的研究一致,GAD酶存在pH作用区间狭窄的问题。在不同pH条件下考察此酶pH稳定性,处理2 h后,LpGAD在pH 3.6−5.0范围内残留酶活力高于80%,当pH高于5.4时,酶活力严重损失,进一步说明了该酶对中性环境不耐受(图 2D)。综合以上LpGAD的酶学特性结果,应对其pH适用范围进行改造,使其更具工业应用价值。
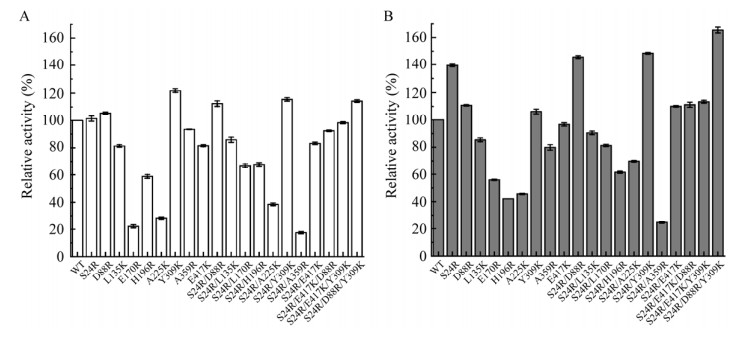
2.1.3 基于表面电荷理性设计突变体及pH特性研究 利用Rosetta Supercharge将酶表面电荷从−76上调到−66,确定9个突变位点,依次构建大肠杆菌突变株,测定其酶学性质。分别以pH 4.5和pH 6.0下野生酶的酶活力值作为100%,比较各单点突变体及组合突变体在对应条件下的相对酶活(图 3),结果显示,除位点H196R、Y309K、A359R外,其余位点在偏中性pH 6.0下活性提高比例皆大于酸性pH 4.5,侧面说明了该理性设计方法具有较好的可行性。单突变点中S24R在pH 6.0下活性提高最明显,Y309K在pH 4.5条件下活性提高最明显。因此,选择了偏中性环境下提高最显著的点S24R继续组合其他位点,结果发现叠加有益单突变位点进一步提升了酶在偏中性环境下的催化能力。同时,组合过程中发现突变体LpGADS24R/A359R较各单突变体并没有表现出活性的提高,这可能是位点组合使酶结合底物与产物的空间结构或活性中心的静电势发生了负向改变,Khersonsky等[36]在对一种小假单胞菌(Pseudomonas diminuta)来源的磷酸三酯酶(phosphotriesterase, PTE)进行突变的过程中,也观察到类似现象。最终得到的三突变体LpGADS24R/D88R/Y309K,在pH 4.5和pH 6.0的酶活分别提高到野生酶对应pH酶活的114%和165%。组合突变得到的正向效果可能是由于不同的残基侧链与催化氨基酸或底物的相互作用造成。
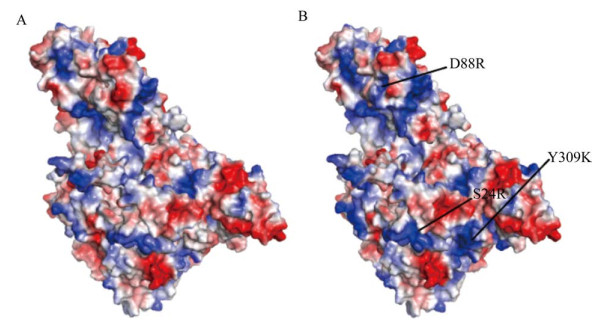
接下来,通过设置不同底物浓度0.1−50 mmol/L,测试了pH 4.5和pH 6.0条件下LpGAD突变前后的动力学变化。用GraphPad Prism 7软件计算反应常数Km和Vmax值,从表 2中可以看出,原始酶LpGAD在pH 4.5下的kcat/Km值高于高pH条件下的值,为2.8 L/(s·mmol),突变酶LpGADS24R/D88R/Y309K为4.5 L/(s·mmol),值得注意的是,虽然突变酶LpGADS24R/D88R/Y309K的kcat/Km值在pH 6.0下小于pH 4.5,但其在pH 6.0时的kcat/Km值显示出较原始酶的绝对优势,是LpGAD对应值的2.3倍。这说明通过累积表面电势变量改变了酶与底物的作用,使突变酶在偏中性pH下拥有了更好的底物催化能力。突变后该蛋白的表面电势图如图 4所示。可以看出,一些显著的酸表面聚集体已被取代。
表 2 原始酶LpGAD和突变酶的动力学参数
Table 2 Kinetic parameters of initial enzyme LpGAD and its mutant towards l-glutamic acid substrate
| Variant |
Km(mmol/L) |
kcat
(1/s) |
kcat/Km
(L/(s·mmol)) |
| LpGAD (pH 4.5) |
8.9 |
24.8 |
2.8 |
| LpGAD (pH 6.0) |
10.6 |
19.5 |
1.8 |
| LpGADS24R/D88R/Y309K (pH 4.5) |
8.3 |
37.6 |
4.5 |
| LpGADS24R/D88R/Y309K (pH 6.0) |
9.3 |
38.2 |
4.1 |
2.2 谷氨酸脱羧酶突变体LpGADS24R/D88R/Y309K的酶学性质研究及动力学解析
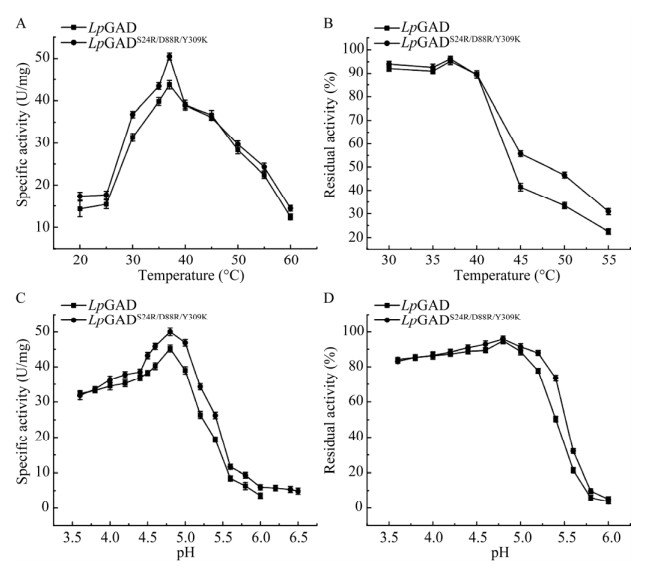
2.2.1 LpGADS24R/D88R/Y309K的酶学性质研究 突变体LpGADS24R/D88R/Y309K在不同温度下的酶活测定如图 5A所示。突变体酶活力曲线整体趋势与野生酶相似,最适温度为37 ℃,整体较野生酶有微弱提升。将突变体与野生酶在各温度条件下保温10 h测定残留活力(图 5B),定义保温前酶活力大小为100%。结果显示,突变体在最适温度附近残留酶活力与野生酶无明显差异,但随着温度的上升(大于40 ℃),其残留活力明显大于野生酶,在50 ℃时,突变体LpGADS24R/D88R/Y309K残留近50%的活性,约为野生酶的1.4倍。突变体在不同pH条件下的比活力大小(图 5C)及在各pH条件下保温2 h后的残留活性曲线(图 5D)显示,突变酶在小于最适pH 4.8条件下,酶活力大小与野生酶差异不明显,但随着pH逐渐上升,突变体LpGADS24R/D88R/Y309K酶活力较野生酶有明显的提升,在pH 6.0时,突变体酶活力是同条件下野生酶活力的168%,且突变体在pH 6.5的条件下仍具有一定的酶活力,改变了野生酶在大于pH 6.0时无活性的特性。当突变体在pH 3.6−5.0下保温2 h后仍能保持90%左右的活力,在pH大于5.0的条件下,突变体残留酶活下降趋势较野生酶有所减缓,其中在pH 5.4下突变体残留活性约为70%,远高于野生酶。以上有关突变体LpGADS24R/D88R/Y309K的酶学性质研究结果,说明了突变不仅拓宽了酶作用pH范围,还对改善酶在较高温及偏中性pH环境下的耐受性有重要影响。
2.2.2 分子动力学模拟解析突变的影响 为了从分子水平了解突变引起的变化,通过分子动力学模拟研究了出发酶LpGAD和突变体LpGADS24R/D88R/Y309K在结合模式以及相关动力学参数上的差异。
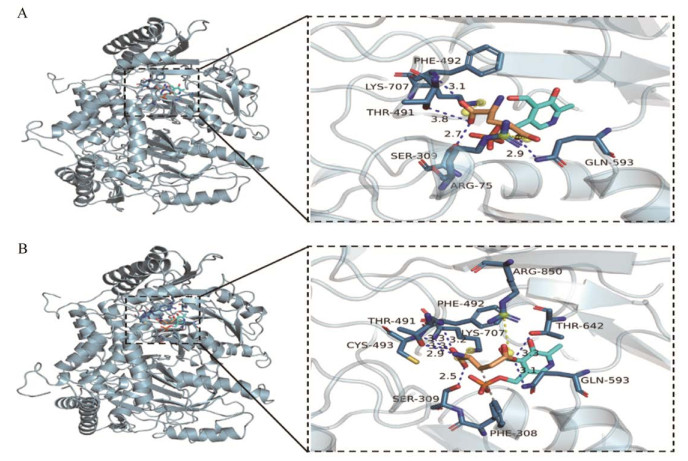
从图 6中可以观察到,谷氨酸与LpGAD的结合通过不同的作用力维持,例如,小分子和蛋白上的Gln-593、Ser-309、Thr-491、Phe-492形成的氢键作用以及和Lys-707、Arg-75形成的盐桥作用。而在谷氨酸与LpGADS24R/D88R/Y309K的复合物中,可以观察到更多的作用力,例如,谷氨酸可以和Thr-642、Cys-493形成额外的氢键作用,和Arg-850形成盐桥作用,以及和Phe-308形成疏水作用。多作用力的形成利于底物的稳定结合。
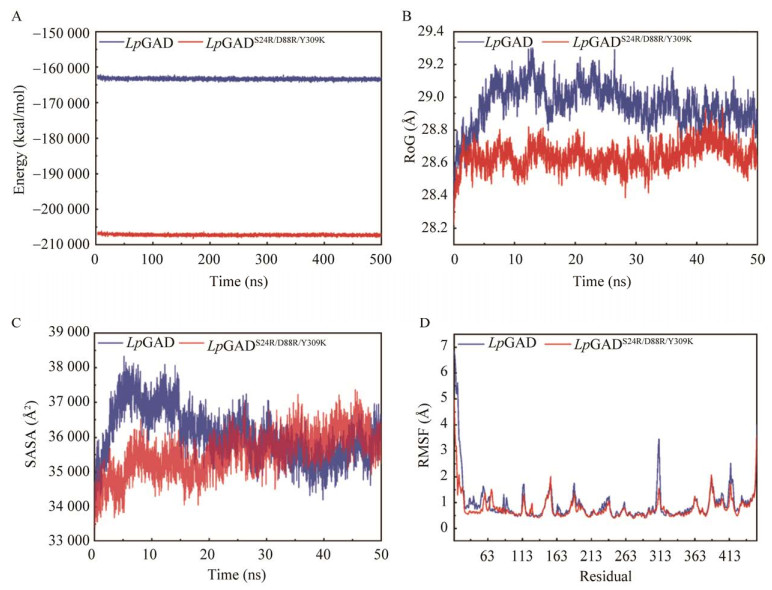
此外,就复合物体系能量、溶剂可及表面积(solvent-accessible surface area, SASA)、旋转半径的时间差(radius of gyration, Rog)和复合物均方根波动(root mean square fluctuation, RMSF)的变化参数分析了突变的影响。从图 7可知,在相同模拟条件下,突变体系的能量低于原体系,平均总能量为−207 000 kcal/mol,较初始值降低了43 000 kcal/mol,这表明突变增加了酶的结构稳定性。RoG反映分子中心与原子质量的关系,可表征蛋白质结构的紧密性。从RoG中可以观察到LpGADS24R/D88R/Y309K体系的RoG值减少,表明酶的构象发生了收缩,暗示突变体系的三元复合物更加密实且稳定。通过分析溶剂可及表面积发现,在模拟前期,2个复合物SASA相当,但是在模拟后期突变体LpGADS24R/D88R/Y309K体系的SASA较小,暗示该突变体系具有内敛的趋势,突变使得体系内在作用增加,即蛋白与底物以及辅因子作用更密切。RMSF通过提供每个氨基酸的运动性信息,反映了模拟过程中蛋白的柔性,可用于分析热稳定性。从图中可以看到两体系的RMSF整体都较小,表明残基的灵活性没有显著改变。但突变体在多处序列位置表现出更低的RMSF,且在氨基酸序号298−328处显著降低了蛋白的柔性,这种蛋白刚性的增加可能会对底物结合产生影响,利于稳定结合小分子底物,降低底物“脱靶”的敏感性,从而更好地催化反应发生。
2.3 全细胞转化体系的优化
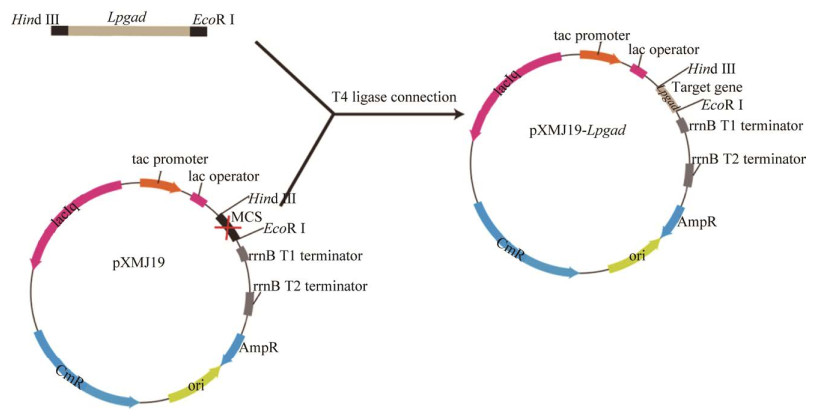
2.3.1 重组谷氨酸棒杆菌的构建及诱导表达 谷氨酸棒杆菌作为食品安全菌株GRAS,培养简单,不产内毒素,适用于原核生物基因的克隆表达及重组蛋白的有效分泌,在多种氨基酸及其衍生物生产中被广泛利用[23]。由方法1.2.3构建表达Lpgad基因及LpgadS24R/D88R/Y309K突变基因的谷氨酸棒杆菌重组菌株C. glutamicum E01/pXMJ19-Lpgad和C. glutamicum E01/pXMJ19-LpgadS24R/D88R/Y309K。质粒构建流程如图 8所示,重组谷氨酸棒杆菌转化子质粒测序验证成功后,在BHI培养基中逐步扩大培养。50 mL体系培养5 h后加入IPTG诱导蛋白表达,20 h后离心收集菌体,进行摇瓶转化。
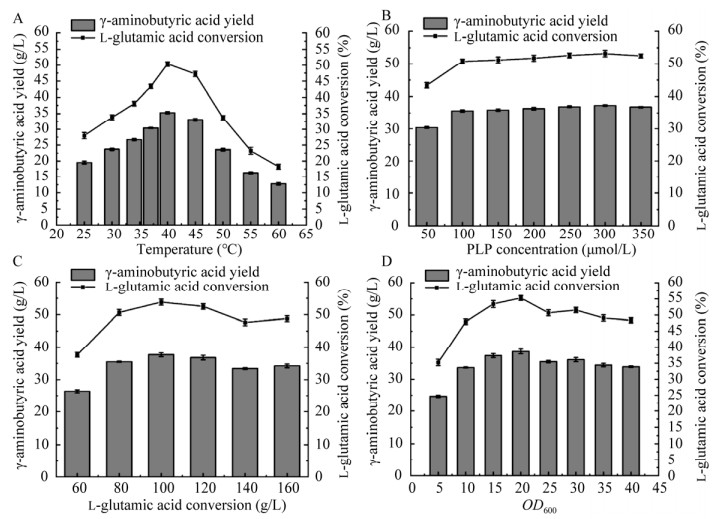
2.3.2 温度对全细胞转化合成GABA的影响 转化温度通过影响菌体的生长状态以及酶的催化效率间接影响GABA的合成。考察了不同温度25−60 ℃对全细胞催化合成GABA的影响。结果如图 9A所示,在反应温度为40 ℃时GABA的产量最高,为35.3 g/L。当温度大于50 ℃时,GABA的产量下降明显,说明高温会影响突变酶LpGADS24R/D88R/Y309K的活力,降低酶催化效率。
2.3.3 辅助因子浓度对全细胞转化合成GABA的影响 在全细胞转化中,GAD酶依赖少量PLP发挥催化作用,适当浓度的PLP能够促进重组菌合成产物,使GABA产量不断提高,所以需要对PLP的添加量进行优化。在最适转化温度40 ℃条件下,设置PLP浓度梯度为50−350 μmol/L,进行全细胞转化。如图 9B所示,随着PLP添加量的增加,GABA的转化率不断增加。当PLP添加浓度为100 μmol/L时,GABA产量为35.6 g/L。当添加浓度≥100 μmol/L时,GABA产量上升幅度减缓,表明辅酶添加浓度≥100 μmol/L时就可以满足反应的需求,出于经济性原则考虑,选取PLP浓度100 μmol/L进行后续优化实验。
2.3.4 底物浓度对全细胞转化合成GABA的影响 适当的底物浓度有利于促进产物的高效合成,控制转化温度40 ℃,辅酶PLP浓度100 μmol/L,设置底物谷氨酸浓度范围60−160 g/L,进行全细胞转化,以确定最优底物浓度。结果如图 9C所示,在l-谷氨酸浓度为100 g/L时,GABA的产量最高,为37.7 g/L,选取底物浓度100 g/L进一步优化。
2.3.5 菌体量对全细胞转化合成GABA的影响 在实际工业生产中,控制反应过程中的菌体量能有效降低成本和提高产率。设置菌体量OD600为5−40的不同梯度,在上述最适条件即反应温度40 ℃,PLP浓度100 μmol/L,谷氨酸浓度100 g/L下进行全细胞转化。结果如图 9D所示,在菌体OD600=20时,GABA的产量最高,为38.7 g/L。
2.4 5 L发酵罐水平全细胞分批补料转化合成GABA 为了实现GABA的放大生产,研究了重组谷氨酸棒杆菌C. glutamicum E01/pXMJ19-Lpgad和C. glutamicum E01/pXMJ19-LpgadS24R/D88R/Y309K在5 L发酵罐水平上GABA的合成情况。用0.9%的NaCl溶液为缓冲体系分别悬浮了C. glutamicum E01/pXMJ19-LpgadS24R/D88R/Y309K和C. glutamicum E01/pXMJ19-Lpgad菌体,定容至1 L,控制初始反应条件一致,菌体量OD600= 20,底物浓度100.0 g/L,PLP浓度100 μmol/L,转化温度40 ℃,于5 L发酵罐中进行转化,转速300 r/min,不控制pH,14 h内分批次投加100 g底物l-谷氨酸。各时间段取样测得转化液中产物含量及底物谷氨酸的含量如图 10所示。随着转化的进行,底物消耗速度减缓,依次增大投料时间间隔,重组菌C. glutamicum E01/pXMJ19-LpgadS24R/D88R/Y309K在14 h内实现9次投料,考虑投料体积引起的变化,测得20 h时GABA产量高达402.8 g/L,前期转化率可达95.5%。C. glutamicum E01/pXMJ19-Lpgad菌株14 h内实现6次投料,20 h时GABA积累量为247.1 g/L。同时检测各时期转化液pH值发现,在20 h时,C. glutamicum E01/pXMJ19-LpgadS24R/D88R/Y309K转化液pH值达到6.1,高于C. glutamicum E01/pXMJ19-Lpgad转化液pH值。以上结果表明,通过提高LpGAD酶在偏中性pH下的催化效率,能够提高制备γ‑氨基丁酸的转化效率,缩短生产周期。
3 讨论与结论 本研究基于表面电荷的策略理性设计LpGAD酶,通过在谷氨酸棒杆菌中异源表达优势突变酶LpGADS24R/D88R/Y309K,实现了全细胞催化l-谷氨酸高效合成γ-氨基丁酸。首先从实验室保存的一株ARTP诱变后的高产GABA的植物乳杆菌中克隆表达GAD酶,并测定其酶学性质,发现其最适pH偏酸性,但在GABA转化生成过程中,随着GABA生成,溶液pH会逐渐上升,不利于GAD酶的持续转化,因此,有必要拓宽其pH适用范围,以实现GABA的高效合成。本研究采用Rosetta Supercharge理性设计LpGAD,共选取9个突变位点进行定点突变,获得的三突变体LpGADS24R/D88R/Y309K成功提升了最适pH下的活力,同时提升了酶在偏中性pH下的酶活,通过酶学性质研究及动力学模拟进一步分析突变的影响。接着将LpGAD和LpGADS24R/D88R/Y309K在C. glutamicum E01中进行过表达构建单细胞工厂,为进一步提高全细胞催化体系的效率,从温度、PLP辅助因子浓度、底物浓度和菌体量4个方面优化了全细胞催化体系,确定最适转化条件为反应温度40 ℃,PLP浓度100 μmol/L,菌体量OD600为20,底物谷氨酸浓度为100.0 g/L。最终的工程菌C. glutamicum E01/pXMJ19-LpgadS24R/D88R/Y309K在最适反应条件下,于5 L罐放大转化体系中,初始添加1 L转化液,14 h内分10批次共投料1 000 g谷氨酸高效合成γ-氨基丁酸,产量高达402.8 g/L,前期摩尔转化率为95.5%,达到国内领先水平。目前,国内Wen等[37]以葡萄糖和玉米秸秆水解液为底物利用重组谷氨酸棒杆菌发酵合成γ-氨基丁酸,发酵72 h和40 h,产量达77.6 g/L和39.2 g/L,得率达到0.44 g/g葡萄糖和0.39 g/g葡萄糖;Ke等[38]以过表达乳酸乳球菌(Lactococcus lactis) FJNUGA01来源的GAD酶的重组大肠杆菌为生产菌,在条件优化后全细胞催化生产γ-氨基丁酸,7 h后最终产物浓度约204 g/L,产物产量较低。本研究一定程度上解决了γ-氨基丁酸全细胞催化生产过程中关键酶pH作用区间较小的问题,不仅提升了LpGAD酶在最适pH下的酶活,同时提升了酶在偏中性pH下的酶活,从而增强了该酶合成γ-氨基丁酸的能力,使其更适应于工业生产。同时,本研究也对其他生产用关键酶pH改造提供了一定指导意义。
 2023, Vol. 39
2023, Vol. 39