1. 浙江科技学院生物与化学工程学院, 浙江 杭州 310023;
2. 浙大宁波理工学院生物与化学工程学院, 浙江 宁波 315100;
3. 浙江大学化学工程与生物工程学院, 浙江 杭州 310027;
4. 金华高等研究院, 浙江 金华 321019
收稿日期:2022-11-18;接收日期:2023-01-26;网络出版时间:2023-01-31
基金项目:国家自然科学基金(32071268);宁波市“科技创新2025”重大专项(2020Z080)
作者简介:邱帅 浙江科技学院生物与化学工程学院讲师,主要研究领域为生物酶催化、蛋白质工程,在Chemical Engineering Science、Bioorganic Chemistry等期刊发表SCI论文共计13篇,其中第一作者5篇,申请发明专利6项,获专利授权3项,近五年主持国家自然科学青年基金项目1项,参与国家自然科学基金面上项目2项;
黄俊 浙江科技学院教授、博士生导师、生物与化学工程学院院长。已主持国家自然科学基金4项、浙江省重大科技专项2项、企业重大横向多项,在国际学术期刊Biotechnol Bioeng、Biochemistry等发表学术论文60余篇,获得国家市场监管科研成果奖一等奖1项、轻工业联合会科学技术进步奖二等奖1项。入选浙江省高校领军人才(高层次拔尖人才)、浙江省"新世纪151人才工程"第二层次培养人员,曾获2020年浙江省抗击新冠疫情先进个人、浙江省第五届师德先进个人、浙江省科技帮扶促调先进个人.
1. School of Biological and Chemical Engineering, Zhejiang University of Science and Technology, Hangzhou 310023, Zhejiang, China;
2. School of Biological and Chemical Engineering, Ningbo Tech University, Ningbo 315100, Zhejiang, China;
3. College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, Zhejiang, China;
4. Jinhua Advanced Research Institute, Jinhua 321019, Zhejiang, China
Received: November 18, 2022; Accepted: January 26, 2023; Published: January 31, 2023
Supported by: This work was supported by the National Natural Science Foundation of China (32071268) and the Ningbo "Scientific and Technological Innovation 2025" Key Project (2020Z080)
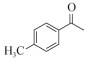
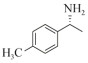
手性胺是手性中心含有氨基的一类小分子化合物,它是多种药物活性分子的重要结构单元[1-3]。据统计,在最常用的200种小分子处方药中,超过1/3含有手性胺结构[4-5],如用于治疗阿尔兹海默病的卡巴拉汀[6]、抗糖尿病药物的主要成分西他列汀[7]、拟钙剂盐酸西那卡塞[8]等。酶促不对称合成是制备手性胺的重要方法,常用的酶有单加氧酶、亚胺还原酶、氨基酸脱氢酶、转氨酶等[9-13]。其中ω-转氨酶因底物谱相对广泛、辅酶相对廉价、产物纯度高等优点,受到研究人员广泛青睐[14-15]。
ω-转氨酶(ω-transaminase, ω-TA)是一种5′-磷酸吡哆醛(pyridoxal 5′-phosphate, PLP)依赖性酶[16-17],以酮类化合物为原料,将供体分子中的氨基立体选择性地转移到前手性酮分子上,高效制备手性胺[5, 18],反应过程如图 1所示。目前,已在多种微生物中发现了ω-转氨酶,包括河流弧菌(Vibrio fluvialis)、节杆菌(Arthrobacter)、巨大芽孢杆菌(Bacillus megaterium)和土曲霉(Aspergillus terreus)等[19-22]。虽然ω-转氨酶在合成手性胺方面有较好的应用前景,但目前发现的野生酶存在稳定性差、催化效率低等诸多不足,不利于应用到工业生产中[23]。此外,在酶催化过程中,提高反应温度可以提高疏水性底物的溶解度[24]、防止微生物的生长[25]、增加反应速率[26]等,然而大多数酶喜室温或低温条件[27],难以抵抗高温反应条件,因此,对酶的热稳定性进行改造具有重要的应用价值。蛋白质工程作为强有力的工具在酶分子改造中愈发重要,通过蛋白质工程改善酶的热稳定性具有重要的科学意义[28-29]。
在酶的热稳定性改造的过程中,普遍存在稳定性与活性之间相互权衡的trade-off效应[30-33],建立一种高效提升热稳定性而不降低酶活性的方法仍然是一重要挑战。Cao等[34]采用随机-半理性相结合的突变策略对节杆菌属来源的ω-转氨酶进行改造,最终获取了稳定性与活性同步提升的突变体;Jia等[35]对毛霉属来源的转氨酶进行口袋改造,最终获得同步进化的鲁棒性酶。
本研究通过对来自土曲霉属的ω-转氨酶(AtTA)进行不同温度下的分子动力学模拟锚定影响酶热稳定性的关键氨基酸区域,采用随机突变与组合突变相结合的策略,获得热稳定性和活性同时提高的突变酶,进而从分子机制解释酶稳定性与活性同步提升的原因。
1 材料与方法
1.1 材料和试剂 菌株与质粒:表达宿主大肠杆菌(Escherichia coli) BL21(DE3)购自北京全式金生物技术有限公司、重组质粒pET-28a(+)-AtTA由安徽通用生物公司构建并合成。
Luria-Bertani (LB)培养基:称取10.0 g蛋白胨、10.0 g氯化钠、5.0 g酵母粉溶于去离子水中并定容至1 L,121 ℃灭菌20 min。
酶、试剂、引物和脱氧核糖核酸(deoxyribonucleic acid, DNA)序列测定:限制性核酸内切酶和胶回收试剂盒购自TaKaRa公司;Bradford蛋白浓度测定试剂盒购自生工生物工程(上海)股份有限公司;引物合成和DNA测序由有康生物科技有限公司完成。
1.2 易错PCR构建随机突变文库 以pET-28a(+)-AtTA为模板,使用引物P1、P2、P3和P4 (表 1)进行目的片段扩增。在PCR反应混合体系中加入100 μmol/L MnCl2水溶液用于控制突变概率,使得每个基因有1−3个突变位点。以扩增后的目的片段为引物,以pET-28a(+)-AtTA为模板进行全质粒PCR。将PCR产物回收纯化并使用Dpn Ⅰ处理以切除模板DNA,然后加入到E. coli BL21(DE3)感受态细胞中,混合冰浴30 min,42 ℃水浴热击90 s,置于冰上冷却5 min,再向eppendorf管中加入650 μL LB液体培养基,37 ℃、180 r/min培养2 h。取100 μL菌液均匀涂布于含有50 μg/mL的硫酸卡那霉素的LB平板上,37 ℃培养24 h。
表 1 易错PCR和定点突变所使用的引物列表
Table 1 Primers for error-prone PCR and site- directed mutagenesis
| Primer name |
Primer sequence (5′→3′) |
| P1-F |
CTGGAAAGCACCGAAACCACCAACCCGTTT |
| P1-R |
CCGAGCGTGTGGGATGGCCGCTTTTTTCGC |
| P2-F |
CATATTACCCGCCTGGAAGCGAGCTGCACC |
| P2-R |
GGCGTGCGCGGCACCCGCCCGGAAGATATT |
| P3-F |
AGCGGCTTTAACATTGTGCTGGTGAAAGAT |
| P3-R |
GAAATTTTTATGTGCACCACCGCGGGCGGC |
| P4-F |
GGCGTGACCCGCAAAAGCGTGATTAACGCG |
| P4-R |
GGCATGCCGGTGAACGGCGGCCAGATTGGC |
| A42H-F |
GTTCCTTTACATGAAGCACGC |
| A42H-R |
GCGTGCTTCATGTAAAGGAAC |
| E104D-F |
ATCCTGGTGGATATGGTCGCA |
| E104D-R |
TGCGACCATATCCACCAGGAT |
| A246V-F |
GTTATCAACGTTGCTGAAGCC |
| A246V-R |
GGCTTCAGCAACGTTGATAAC |
| R266Q-F |
GGCCTACCAGTGTGACGAGATT |
| R266Q-R |
AATCTCGTCACACTGGTAGGCC |
| The underlined codons are amino acid codons after the mutation. |
1.3 高通量筛选方法 使用无菌牙签挑取平板上的单菌落接种至含1 mL LB培养基(含50 μg/mL硫酸卡那霉素)的96深孔板中,于37 ℃、180 r/min培养8 h后加入终浓度为0.5 mmol/L的异丙基-β-d-硫代半乳糖苷(isopropyl β-d-1-thiogalactopyranoside, IPTG),在25 ℃、150 r/min培养18 h诱导目的蛋白表达。诱导结束后,在4 500 r/min离心15 min收集菌体,将菌体用磷酸盐缓冲液(50 mmol/L, pH 8.0)重悬离心洗涤2次后置于–80 ℃冷冻20 h,反复冻融3次后向每孔中加入250 μL细胞裂解液(5 mg/mL溶菌酶),重悬菌体后于37 ℃、400 r/min孵育1 h,4 ℃、4 500 r/min离心15 min收集上清。转移各孔100 μL上清至96微孔板的相应位置,将96微孔板置于35 ℃恒温仪中孵育30 min,随后将96微孔板冰浴10 min。吸取20 μL冰浴处理后的酶溶液置于石英96孔板中,加入180 μL反应溶液[50 mmol/L pH 8.0的磷酸盐缓冲液,含0.25% (体积分数)二甲基亚砜(dimethyl sulphoxide, DMSO)、2.5 mmol/L丙酮酸、2.5 mmol/L (R)-(+)-苯乙胺、0.1 mmol/L PLP],在30 ℃、pH 8.0条件下反应3 min后,用酶标仪检测波长在245 nm处相应的酶活[36]来初筛正突变子。
1.4 组合突变体的构建及筛选 将通过易错PCR和高通量筛选获得的突变体进行组合,构建多位点突变体。以重组质粒pET-28a(+)-AtTA为模板,采用全质粒PCR方法进行定点突变,经Dpn Ⅰ消化PCR模板后,将构建的质粒转化至大肠杆菌E. coli BL21(DE3),挑选有益突变体用于表达纯化并进行酶的活性测定。通过高效液相色谱(high performance liquid chromatography, HPLC)测定生成的目的手性胺产物丙氨酸的生成量,进而计算酶的活性。一个酶活单位U定义为:在30 ℃、pH 8.0条件下每分钟每生成1 μmol的丙氨酸所需要的酶量。比酶活定义为每毫克蛋白所具有的酶活性单位数,记为U/mg。
丙氨酸HPLC检测方法:向0.2 mL反应后的溶液中加入0.3 mL 200 mmol/L NaHCO3 (pH 9.8)终止反应,然后向其中加入0.5 mL丹磺酰氯溶液(4 mg/mL),在40 ℃条件下进行衍生化1 h,用0.22 μm滤膜将反应液过滤后进行HPLC检测,色谱柱为Agilent InfinityLab Poroshell 120 EC-C18 column (2.1 mm×100 mm, 1.8 μm)。使用由溶液A (水: 甲酸=99.9:0.1, 体积比)和溶液B (乙腈: 甲酸=99.9:0.1, 体积比)组成的流动相进行梯度洗脱。洗脱梯度:溶液B在15 min内从20%逐渐增加到100%,然后在5 min后降至20%,流速为1.0 mL/min,UV (ultraviolet)检测波长为254 nm。
1.5 酶的表达与纯化 挑取野生型及突变体单菌落接种至5 mL含50 μg/mL硫酸卡那霉素的LB培养基中,37 ℃、200 r/min条件下培养8 h后,将菌液转接至200 mL的LB培养基中(含50 μg/mL硫酸卡那霉素),37 ℃、180 r/min培养至OD600为0.6−0.8时,加入终浓度为0.5 mmol/L的IPTG,25 ℃、150 r/min条件下继续培养18 h,于4 ℃、8 000 r/min离心收集菌体。
将收集的菌体经磷酸盐缓冲液(50 mmol/L, pH 8.0)离心洗涤2次后重悬于50 mL细胞破碎缓冲液(50 mmol/L NaH2PO4, 300 mmol/L NaCl, 20 mmol/L咪唑溶液,pH 8.0)中,在75 MPa下高压均质2 min,直至溶液澄清后,4 ℃、8 000 r/min离心50 min,收集上清液。将上清液经0.45 μm滤膜过滤后由Ni-NTA亲和层析获取目标蛋白,依次加入3倍柱体积的细胞破碎缓冲液和清洗缓冲液(50 mmol/L NaH2PO4, 300 mmol/L NaCl, 50 mmol/L咪唑溶液,pH 8.0)除去杂蛋白,随后用1倍柱体积的洗脱缓冲液(50 mmol/L NaH2PO4, 300 mmol/L NaCl, 250 mmol/L咪唑,pH 8.0)收集目的蛋白,并分别采用十二烷基硫酸钠聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis, SDS-PAGE)电泳和改良型Bradford法测定纯化后的蛋白纯度及浓度。
1.6 酶学性质的测定
1.6.1 热稳定性测定 酶的热力学参数表征主要包括半衰期(t1/2)、半失活温度(T1050)和热解折叠温度(Tm)。t1/2是指在特定温度下,酶的活力下降至50%所需的时间。T1050是指在不同温度孵育10 min后,酶的活力降至50%的温度。Tm是指蛋白质在温度上升的过程中,50%蛋白质去折叠时的温度。
t1/2测定:将野生酶和突变酶分别在35 ℃孵育0–180 min或40 ℃孵育0−30 min后立即在冰上冷却10 min,然后取20 μL纯酶稀释液和180 μL的底物溶液在30 ℃、pH 8.0条件混合反应3 min后,通过HPLC检测产物丙氨酸的生成量,并计算相应的酶活力。利用一级失活方程计算半衰期。热失活方程为:
其中kd为酶失活动力学常数,At是不同时间下的酶活,A0为初始酶活,当At为A0的一半时,对应的时间t即为酶的半衰期t1/2。
T1050测定:纯化的酶溶液分别于4、30、35、37、40、42、45、47、50、55 ℃下孵育10 min,然后在冰上冷却10 min,采用上述方法测定酶活力。
Tm测定:示差扫描荧光法(differential scanning fluorimetry, DSF)是一种鉴定蛋白热稳定性的快速高效的方法[37]。荧光染料分子对于蛋白质表面的疏水部分有亲和力,能够在蛋白质解开三级结构时发出荧光信号,从而通过监控荧光染料分子的荧光值随时间逐渐增强来计算蛋白质的热解折叠温度。取49 μL 0.1 mg/mL纯酶稀释液、1 μL 50×SYPRO Orange染料混合后利用荧光定量PCR仪进行测量。扫描温度为25−70 ℃,每次升温0.7 ℃并在该温度保持30 s,激发波长和发射波长分别为490 nm和605 nm。Tm计算公式如下所示:
其中UF和NF分别为最小和最大的荧光发射强度,α是T0内曲线的斜率。
1.6.2 动力学参数的测定 分别设定丙酮酸和1-(R)-苯乙胺的浓度梯度为0−5 mmol/L和0−3 mmol/L,同时固定另一种底物的浓度为2.5 mmol/L,测量不同底物浓度下的反应速率,计算AtTA及其突变体分别对1-(R)-苯乙胺和丙酮酸的动力学参数。动力学结果使用Origin 8.0中的Michaelis-Menten方程进行拟合计算。
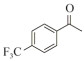
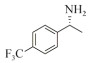
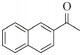
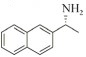
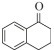
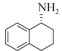
1.7 底物谱测定 以1-(R)-苯乙胺为胺基供体,选定苯甲醛、苯乙酮、4-氟苯乙酮、4-氯苯乙酮、4-溴苯乙酮、4-甲氧基苯乙酮、4-甲基苯乙酮、4-三氟甲基苯乙酮、4-硝基苯乙酮、1-乙酰基萘、2-乙酰基萘、α-四氢萘酮为氨基受体,进行催化反应。2 mL反应体系分别包括10 mmol/L 1-(R)-苯乙胺、10 mmol/L各酮类底物、0.1 mmol/L PLP、50 mmol/L磷酸盐缓冲液(pH 8.0)、0.15 mg/mL纯酶液。30 ℃、180 r/min反应24 h后通过HPLC测定各产物生成量并计算产率与产物的对映体过量值(enantionmeric excesses, e.e.)。
HPLC检测方法:用0.22 μm滤膜将反应液过滤后进行HPLC检测,色谱柱为Agilent InfinityLab Poroshell 120 EC-C18 column (4.6 mm×150 mm, 4.0 μm),流动相为乙腈: 水= 40:60 (体积比),流速为1.0 mL/min,UV检测波长为210 nm。
1.8 分子动力学模拟 从PDB (protein data bank)数据库中搜索得到来源于土曲霉(Aspergillus terreus)的(R)-ω-转氨酶晶体结构(PDB ID: 4CE5)作为模板,采用FoldX建模方法构建突变体的三维结构,并使用YASARA 16.4.6软件(http://www.yasara.org)中的Amber14力场,在303 K和313 K温度下对野生酶和突变酶进行时长为20 ns的动力学模拟[38]。蛋白质的空间环境是充满密度为0.998 g/mL的水的10 Å×10 Å×10 Å立方体,介质中加入Na+和Cl– (0.9%)作为对抗离子形成电中性体系,可电离基团在pH 8.0时根据pKa值质子化。在分子力学水平上,将范德华作用力和静电相互作用的短程截断距离设置为8.0 Å,采用Particle Mesh Ewald方法计算长程静电相互作用。
2 结果与分析
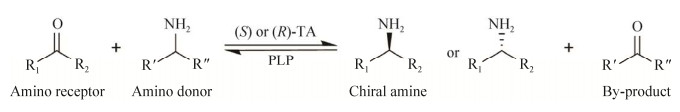
2.1 有义突变区域的鉴定 分子动力学(molecular dynamics, MD)模拟中的均方根波动(root mean square fluctuation, RMSF)值通常反映单个氨基酸残基的波动,对RMSF数值高的氨基酸残基进行突变是常见的提高酶热稳定性的方法。RMSF值与所对应的氨基酸及其区域的柔性呈正相关[39]。对AtTA野生酶分别在35 ℃和45 ℃下进行20 ns的分子动力学模拟,并分析每个氨基酸位点的RMSF偏移值,对35 ℃和45 ℃下氨基酸位点的RMSF值取差值(ΔRMSF=RMSF45 ℃–RMSF35 ℃)得到氨基酸残基随着温度的升高,RMSF变化较大的位点及所在区域。如图 2A、2B所示,氨基酸位点RMSF值变化较大的区域主要集中于4个区域(图中所示Hotspot region 1, Hotspot region 2, Hotspot region 3, Hotspot region 4),通过分析4个热点区所在的蛋白结构位置,我们发现,Hotspot region 2, 3, 4位于α-螺旋区域处,Hotspot region 1则位于α-螺旋、β-折叠、loop区域的交界处(图 2C)。选取这4个潜在的突变热点区域进行AtTA的热稳定性改造,以期获得影响酶热稳定性的关键氨基酸位点。
2.2 随机突变与组合突变构建突变体 采用易错PCR方法对WT的4个突变热点区域进行随机突变,以丙酮酸和1-(R)-苯乙胺作为底物进行高通量筛选,最终筛选出4个热稳定性提升的单突变体:A42H、E104D、A246V、R266Q,其残余活性较WT分别提升了6.5%、10.9%、5.0%、46.3% (图 2D)。将这4个单突变体进行组合,构建了以下8个组合突变体:A42H/E104D、A42H/A246V、A42H/R266Q、E104D/A246V、E104D/R266Q、A42H/E104D/A246V、E104D/A246V/R266Q、A42H/E104D/A246V/R266Q,并对8个组合突变体的热力学、动力学参数等酶学性质进行测定。
2.3 野生酶和突变酶的热稳定性测定 按照1.6.1所述方法测定了野生酶与突变酶的半衰期(t1/2)、半失活温度(T1050)和热解折叠温度(Tm),结果如表 2所示。相比于野生酶,4个单突变体的热稳定性均有不同程度的提高,其中R266Q的热稳定性提升最明显,其在35 ℃下的半衰期由17.8 min提升至83.0 min,是野生酶的4.7倍,半失活温度提升至40.3 ℃,较野生酶(38.1 ℃)提高2.2 ℃。组合突变体中,与R266Q相组合的突变体较野生酶的t1/2 (35 ℃)至少提升2.6倍,这说明266位点是提升该酶热稳定性的一个关键位点。最佳突变体E104D/A246V/R266Q (M3)在35 ℃下的半衰期达到102.7 min,较野生酶WT提高了4.8倍,其T1050和Tm值分别比WT提高4.3 ℃和2.4 ℃。
表 2 野生酶和突变酶的热力学参数
Table 2 Stability of WT and mutants
| Enzyme |
35 ℃ t1/2
(min) |
35 ℃ Δt1/2
(min)a |
40 ℃ t1/2
(min) |
40 ℃ Δt1/2
(min)b |
T1050
(℃) |
ΔT1050
(℃)c |
Tm
(℃) |
ΔTm
(℃)d |
| WT |
17.8±1.1 |
0.0 |
6.9±0.6 |
0.0 |
38.1±0.2 |
0.0 |
41.4±0.2 |
0.0 |
| A42H |
20.0±0.7 |
2.2 |
7.6±0.3 |
0.7 |
38.4±0.3 |
0.3 |
42.0±0.4 |
0.6 |
| E104D |
19.6±0.8 |
1.8 |
7.4±0.7 |
0.5 |
38.4±0.7 |
0.3 |
41.5±0.1 |
0.1 |
| A246V |
19.9±0.9 |
2.1 |
7.4±0.2 |
0.5 |
38.2±0.2 |
0.1 |
41.8±0.3 |
0.4 |
| R266Q |
83.0±2.9 |
65.2 |
13.5±0.4 |
6.6 |
40.3±0.7 |
2.2 |
42.6±0.2 |
1.2 |
| A42H/E104D |
18.0±0.9 |
0.2 |
7.1±0.3 |
0.2 |
39.0±0.2 |
0.9 |
42.3±0.5 |
0.9 |
| A42H/A246V |
20.0±1.3 |
2.2 |
7.5±0.6 |
0.6 |
39.3±0.3 |
1.2 |
42.4±0.4 |
1.0 |
| A42H/R266Q |
96.8±2.7 |
79.0 |
21.7±0.5 |
14.8 |
41.9±0.1 |
3.8 |
43.3±0.2 |
1.9 |
| E104D/A246V |
26.9±1.1 |
9.1 |
8.9±0.8 |
2.0 |
38.4±0.2 |
0.3 |
42.5±0.5 |
1.1 |
| E104D/R266Q |
46.5±1.8 |
28.7 |
11.4±0.9 |
4.5 |
40.1±0.3 |
2.0 |
43.3±0.2 |
2.9 |
| A42H/E104D/A246V |
28.5±1.5 |
10.7 |
9.4±0.4 |
2.5 |
38.8±0.8 |
0.7 |
41.7±0.4 |
0.3 |
| E104D/A246V/R266Q (M3) |
102.7±2.6 |
84.9 |
22.7±0.6 |
15.8 |
42.4±0.4 |
4.3 |
43.8±0.2 |
2.4 |
| A42H/E104D/A246V/R266Q |
86.8±2.8 |
69.0 |
17.1±0.7 |
10.2 |
40.9±0.1 |
2.8 |
43.5±0.3 |
2.1 |
| a: The t1/2 values of mutants compared to WT at 35 ℃; b: The t1/2 values of mutants compared to WT at 40 ℃; c: The T1050 values of mutants compared to WT; d: The Tm values of mutants compared to WT. |
2.4 野生酶和突变酶的动力学参数表征 按照1.6.2所述的方法,分别以丙酮酸和1-(R)-苯乙胺为底物测定野生酶和各突变酶的米氏动力学常数,由表 3可知,野生酶对丙酮酸的催化效率(kcat/Kmpyruvate)为2.17 L/(mmol·s),相较于野生酶,所得到的大部分突变体对丙酮酸的催化效率出现了降低的现象,这说明在热稳定性的改造过程中存在活性-稳定性此消彼长的trade-off效应,而最佳突变体E104D/A246V/R266Q的kcat/Kmpyruvate是野生酶的1.59倍,并未出现活性降低的现象。野生酶对1-(R)-苯乙胺的催化效率(kcat/Km1-(R)-PEA)为2.78 L/(mmol·s),最佳突变体E104D/A246V/R266Q的kcat/Km1-(R)-PEA为4.33 L/(mmol·s),是野生酶的1.56倍,由此表明,突变酶E104D-A246V-R266Q实现了活性-稳定性的同步提升。
表 3 野生酶和突变酶的动力学参数
Table 3 Kinetic parameters of WT and mutants
| Enzyme |
kcatpyruvate
(s–1) |
Kmpyruvate
(mmol/L) |
kcat/Kmpyruvate
(L/(mmol·s)) |
kcat1-(R)-PEA (s–1) |
Km1-(R)-PEA
(mmol/L) |
kcat/Km1-(R)-PEA
(L/(mmol·s)) |
| WT |
0.50±0.01 |
0.23±0.02 |
2.17 |
0.64±0.01 |
0.23±0.03 |
2.78 |
| A42H |
1.45±0.04 |
0.71±0.05 |
2.02 |
0.78±0.08 |
0.23±0.02 |
3.39 |
| E104D |
1.55±0.04 |
0.82±0.07 |
1.88 |
0.72±0.06 |
0.24±0.01 |
2.95 |
| A246V |
0.94±0.07 |
0.54±0.06 |
1.74 |
0.76±0.02 |
0.21±0.01 |
3.66 |
| R266Q |
1.77±0.01 |
0.68±0.05 |
2.60 |
0.78±0.04 |
0.26±0.01 |
3.00 |
| A42H/E104D |
1.50±0.08 |
0.69±0.05 |
2.15 |
0.61±0.05 |
0.20±0.01 |
3.00 |
| A42H/A246V |
1.00±0.01 |
0.46±0.02 |
2.17 |
0.61±0.07 |
0.20±0.01 |
3.01 |
| A42H/R266Q |
1.93±0.05 |
0.65±0.01 |
2.97 |
0.74±0.08 |
0.23±0.02 |
3.22 |
| E104D/A246V |
1.05±0.04 |
0.46±0.03 |
2.28 |
0.74±0.05 |
0.20±0.03 |
3.69 |
| E104D/R266Q |
1.03±0.04 |
0.57±0.05 |
1.81 |
0.79±0.05 |
0.25±0.03 |
3.07 |
| A42H/E104D/A246V |
1.00±0.02 |
0.72±0.03 |
1.39 |
0.85±0.02 |
0.33±0.01 |
2.55 |
| E104D/A246V/R266Q (M3) |
1.56±0.03 |
0.45±0.01 |
3.47 |
1.04±0.08 |
0.24±0.01 |
4.33 |
| A42H/E104D/A246V/R266Q |
1.38±0.02 |
0.55±0.01 |
2.51 |
0.76±0.05 |
0.25±.02 |
2.94 |
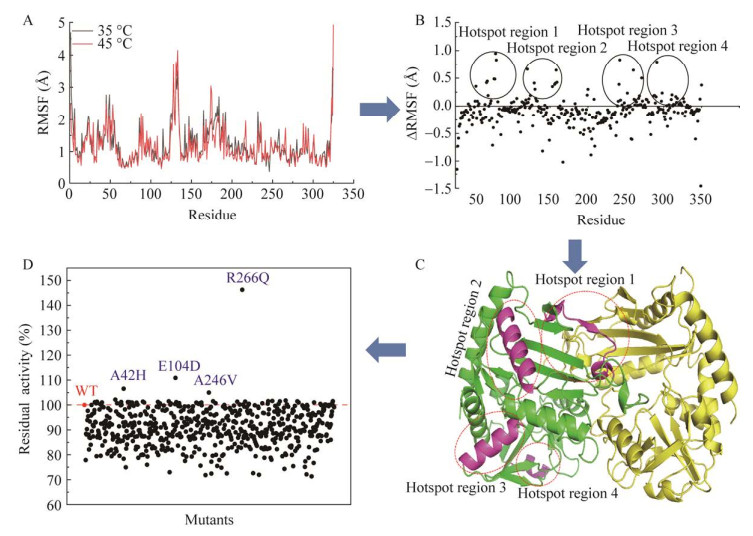
2.5 热稳定性-活性提升的分子机制解析 为阐明突变体M3稳定性与催化效率同步提高的分子机制,通过YASARA进行了分子对接和分子动力学模拟。首先利用FoldX软件以WT晶体结构(PDB: 4CE5)为模板,对M3进行3D结构的重建,并分别对WT和M3进行20 ns的分子动力学模拟,如图 3A所示,M3在5 ns处的结构动态趋于平稳,而WT在7.5 ns时结构趋于平稳,且M3整体的均方根偏差(root mean square deviation, RMSD)值要低于WT,此结果表明M3较WT的蛋白结构更加稳定。进一步分析WT与M3的RMSF值,如图 3B所示,AtTA-M3所编码的325个氨基酸的平均RMSF值(1.154 Å)低于AtTA-WT所编码的RMSF值(1.232 Å)。通过分析3个突变位点104、246、266及其所在的二级结构区域的RMSF值,可以看出,突变体M3中的突变位点及其所在区域的RMSF值均低于WT中位点及所在区域的RMSF值,这一结果表明M3的整体及突变区域的刚性增强,加固了突变酶的热稳定性。
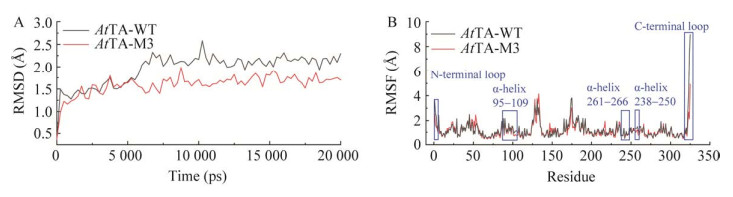
为了更加深入地解释突变位点刚性增强的原因,对突变体M3的突变位点与其周围的氨基酸残基进行了相互作用力的分析,如图 4所示,当104号位点由谷氨酸突变为天冬氨酸时,M3中增加了Asp104与Arg324之间的氢键(图 4A、4D);当246位点的丙氨酸突变为缬氨酸时,Val246与Phe250之间形成1个Pi-烷基作用力(图 4B、4E);当266位点的精氨酸突变为谷氨酰胺时,除了Gln266与Leu263之间存在氢键外,额外增加了Gln266与Glu262之间的氢键(图 4C、4F)。所增加的分子内的相互作用力对突变体M3稳定性的提升具有重要作用。
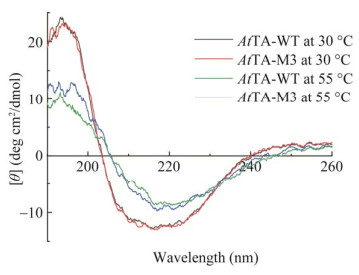
通过对AtTA的三维结构进行分析,我们发现最佳突变体M3的3个突变位点均位于α-螺旋,利用圆二色光谱仪(circular dichroism spectrometer, CD)在190−260 nm波长下的紫外吸收光值对WT及M3的二级结构各元素进行分析,如图 5所示,当温度从30 ℃增加到55 ℃时,WT和M3所保留的二级结构明显减少。在30 ℃和55 ℃下,WT和M3的二级结构表现出差别。随着温度的升高至55 ℃时,WT在平面偏振波长222、208、192 nm下所对应的摩尔椭圆率[θ]分别是–8.45、–2.18、9.41 deg cm2/dmol,M3在3处平面偏振波长下所对应的摩尔椭圆率分别为–9.04、–3.39、11.45 deg cm2/dmol,M3较WT保留下了更多的α-螺旋结构。这表明M3中的α-螺旋更加稳定,这与M3热稳定性的提升也是相对应的。
为了阐明突变体M3活性提升的原因,对WT与M3分别进行与底物丙酮酸的分子对接,作用力模拟分析表明(图 6A),WT与丙酮酸的对接结果显示,丙酮酸与WT的5个氨基酸及PLP之间分别存在2组氢键与4组疏水相互作用力;而突变体M3与丙酮酸之间的相互作用力显示,丙酮酸与M3中6个氨基酸及PLP之间存在4组氢键及4组疏水相互作用力。通过对WT与M3进行底物结合口袋体积计算发现(图 6B),突变体M3较WT的底物结合口袋体积增大,由796.6 Å3 (WT)增加至939.1 Å3 (M3),这一结果表明酶底物结合口袋的扩大,增加了其与底物间碰撞频率,进一步解释了突变酶活力提升的原因。
2.6 野生酶与突变体M3的底物特异性分析 为了验证突变酶M3对不同底物的催化性能,在10 mmol/L各底物浓度下,进行WT和M3的酶催化反应,结果如表 4所示。无论在30 ℃还是40 ℃条件下,反应24 h后,M3对所催化的11种底物的产率较WT均表现出提升的趋势,除S1生成相应的无手性产物外,其他所生成的产物均为严格的R-构型(e.e. > 99.5%)。在30 ℃反应条件下,M3催化11种底物生成相应产物的产率较WT约提升11.1%−229.2%,在40 ℃反应条件下,M3催化11种底物的产率较WT约提升3.7%−125.8%。M3稳定性的提升对于催化酮类底物生成胺产物也表现出明显的优势。总体来说M3对多种芳香酮类底物较WT表现出较好的催化活性。
3 结论 本研究采用随机突变和半理性设计相结合的策略,对来源于Aspergillus terreus的ω-转氨酶AtTA进行蛋白质工程改造,筛选得到热稳定性提升的突变酶R266Q、A42H/R266Q、E104D/A246V/R266Q、A42H/E104D/A246V/R266Q,其中E104D/A246V/R266Q (M3)提升最明显,与WT相比,其t1/2 (35 ℃)由17.8 min提升至102.7 min,提升了4.8倍,且最佳突变体的催化效率略有提升,其kcat/Kmpyruvate、kcat/Km1-(R)-PEA分别是WT的1.59倍、1.56倍,实现了稳定性与活性的同步提升。通过YASARA进行分子对接和动力学模拟分析,α-螺旋结构的加固、底物分子丙酮酸与周围氨基酸氢键的增加以及底物结合口袋体积的增大是M3较WT稳定性和活性提升的原因。M3也较WT对多种芳香酮类化合物表现出更加优良的催化效果,这一结果表明M3在合成多种手性胺类化合物的应用方面具有更大潜力。
 2023, Vol. 39
2023, Vol. 39