L-谷氨酸是一种重要的氨基酸,每年产量占据全球氨基酸市场的400多万t。L-谷氨酸不仅应用于制药业,在食品、生化和畜牧行业也广泛使用[1],目前焦谷氨酸钠(pyroglutamic acid sodium, PCA-Na)、聚谷氨酸等高值产品已成为研究热点[2],因此选育L-谷氨酸高产菌株十分有必要。L-谷氨酸主要通过微生物发酵生产,其在谷氨酸棒杆菌(Corynebacterium glutamicum)内合成途径如图 1所示。谷氨酸棒杆菌是一种食品安全级菌株,最初是由日本研究人员分离出来的一种产生谷氨酸的微生物,具有优秀的大量生产氨基酸的能力,已经广泛应用于各种氨基酸的生产中[3]。谷氨酸棒杆菌在正常生长条件下不能产生谷氨酸,在生物素亚适量或通过添加吐温40或青霉素的情况下可产生谷氨酸[4]。实验室前期通过多级诱变等技术,以谷氨酸中产菌谷氨酸棒杆菌(Corynebacterium glutamicum) E01作为底盘细胞筛选出了一株谷氨酸产量较高的菌株C. glutamicum G01,其产量约为96 g/L,作为宿主菌有很大生产L-谷氨酸的潜力。
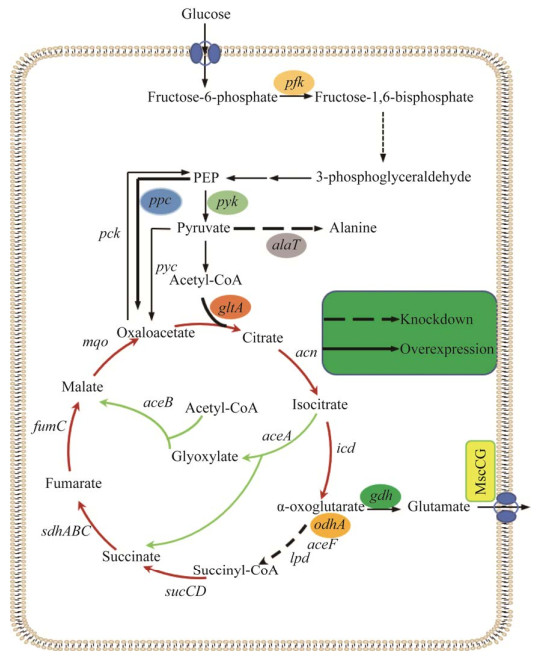
在谷氨酸棒杆菌中,三羧酸(tricarboxylic acid, TCA)循环是产生L-谷氨酸的关键途径。研究表明,α-酮戊二酸脱氢酶复合体(α-ketoglutarate dehydrogenase complex, ODHC)的活性对谷氨酸的合成至关重要,在谷氨酸高产菌中ODHC的活性普遍较低。ODHC由3个亚基组成(E1、E2和E3亚基),其中E1亚基由odhA基因编码。ODHC能够催化α-酮戊二酸生成琥珀酰辅酶A,因此ODHC活性的降低能够使TCA循环更多地向合成谷氨酸流分布,从而提高了谷氨酸的产量[5]。磷酸烯醇式丙酮酸羧化酶(phosphoenolpyruvate carboxylase, PEPC),由ppc基因编码,在TCA循环中起关键作用,能够加强进入TCA循环的碳代谢[6-7]。柠檬酸合酶(citrate synthase, CS),由gltA基因编码,是TCA循环中的第一个酶,参与TCA循环的起始反应,过表达CS可以加强进入TCA循环的碳流量,可能对L-谷氨酸的合成有促进作用[8]。磷酸果糖激酶(phosphofructokinase, PFK)由pfk基因编码,催化果糖-6-磷酸生成果糖-1, 6-二磷酸,其酶活力水平严格影响着糖酵解(Embden-Meyerhof-Parnas, EMP)途径的速率[9];丙酮酸激酶(pyruvate kinase, PYK)由pyk基因编码,控制着丙酮酸的外流量,并在EMP途径中起关键作用,有研究表明过表达PYK能够提高细菌内谷氨酸含量[10]。此外,提高α-酮戊二酸(α-ketoglutaric acid, α-KG)向谷氨酸转化的能力对谷氨酸的合成十分关键,因此选育高性能的谷氨酸脱氢酶(glutamate dehydrogenase, GDH)非常有必要。最后,作为谷氨酸合成的非内源因素,谷氨酸转运蛋白(mechanical sensitive channel of Corynebacterium glutamicum, MscCG)近年来成为研究热点,有研究表明MscCG的结构改变可引起谷氨酸的过量排泄[11]。
本文以实验室前期筛选到的C. glutamicum G01为出发菌株,与前期实验室对C. glutamicum G01发酵前后关键基因转录水平差异的研究不同[12],通过对C. glutamicum E01与C. glutamicum G01进行转录组与基因组测序分析,期望寻找到谷氨酸合成关键途径中基因转录水平的差异,对其合成L-谷氨酸途径进行代谢改造,研究了PEPC和CS对代谢途径的影响,同时结合核糖体结合位点优化以及基因敲除加强了谷氨酸的积累,减少了副产物的生成,强化了谷氨酸代谢流并提高了谷氨酸的外排能力,提高了谷氨酸的产量以及发酵糖酸转化率,为谷氨酸棒杆菌的代谢改造提供了理论基础。
1 材料与方法
1.1 材料
1.1.1 菌株、质粒及培养基 谷氨酸中产菌C. glutamicum E01、谷氨酸高产菌C. glutamicum G01、大肠杆菌(Escherichia coli, E. coli) BL21(DE3)和质粒pXMJ19、pK18mobsacB、pEC-XK99E均为实验室保藏。
LB培养基(g/L):酵母粉5,蛋白胨10,氯化钠10,固体培养基添加1.5%–2.0%琼脂,根据需要添加相应浓度的抗生素。
BHI培养基(g/L):脑心浸液肉汤粉38.5,固体培养基添加1.5%–2.0%琼脂,根据需要添加相应浓度的抗生素。
发酵种子培养基(g/L):葡萄糖25,K2HPO4 1.5,MgSO4 0.6,玉米浆30,FeSO4∙7H2O 0.005,MnSO4∙H2O 0.005,尿素2.5 (与其他成分分开灭菌115 ℃,20 min),pH 7.0。
发酵培养基(g/L):葡萄糖140,K2HPO4 1,MgSO4 0.6,玉米浆5,FeSO4∙7H2O 0.005,MnSO4∙H2O 0.005,尿素7 (与其他成分分开灭菌115 ℃,20 min),pH 7.0。
1.1.2 引物 本研究所用引物如表 1所示。
表 1 本研究所用引物
Table 1 Primers used in this study
| Primers |
Sequences (5ʹ→3ʹ) |
| ΔalaT-1-F |
TATGACATGATTACGAATTCCGATCTGGCTGAGAGAAAAGTAGACA |
| Δ alaT-1-R |
CCCATCCACTAAACTTAAACAATGTTGTGTCCGTCGAGCT |
| Δ alaT-2-F |
TGTTTAAGTTTAGTGGATGGGACCCGAACGTGTACGAAATCC |
| Δ alaT-2-R |
CGACGGCCAGTGCCAAGCTTTGAAGCCTAACCCAGAATAGAGGAC |
| Δ odhA-1-F |
GGAAACAGCTATGACATGATTACGAATTCATCTCACGACGCGTTGTT |
| Δ odhA-1-R |
CCCATCCACTAAACTTAAACATAGCCAATGATGTGGGT |
| Δ odhA-2-F |
TGTTTAAGTTTAGTGGATGGGCCCCGAAGTCCATGCTGC |
| Δ odhA-2-R |
TTGTAAAACGACGGCCAGTGCCAAGCTTTGATCAAGTTCTGTGATGATCTCCTGC |
| p19-gltA-F |
AAACAGAATTAATTAAGCTTAAAGGAGGGAAATCATGTTTGAAAGGGATATCGTGGCT |
| p19-gltA-R |
AAAACAGCCAAGCTGAATTCTTAGCGCTCCTCGCGAGGAACAA |
| RBS1odhA-F |
AAACAGAATTAATTAAGCTTCTCCGCGTCTTACAGCTAGTTTTAACGTGAGCAGCGCTAGTACT |
| RBS2odhA-F |
AAACAGAATTAATTAAGCTTCTCCGCGTCTTACATCTAGTTTTAACGTGAGCAGCGCTAGTACT |
| RBS3odhA-F |
AAACAGAATTAATTAAGCTTCTCCTCGTCTTACAGCTCGTTTTAACGTGAGCAGCGCTAGTACT |
| RBS4odhA-F |
AAACAGAATTAATTAAGCTTCTCCTCTTCTTACAGCTAGTTTTAACGTGAGCAGCGCTAGTACT |
| p19-ppc-F |
AAACAGAATTAATTAAGCTTAAAGGAGGGAAATCATGACTGATTTTCTACGCGATGA |
| p19-ppc-R |
CAAAACAGCCAAGCTGAATTCCTAGCCGGAGTTGCGCAGTGCAGT |
| p19-MscCG-F |
ATTAATTAAGCTTAAAGGAGGGAAATCATGATTTTAGGCGTACC |
| p19-MscCG-R |
CAAAACAGCCAAGCTGAATTCCTAAGGGGTGGACGTTGGCGCAA |
| p19-MscCG-A100V-F |
GCGATTCCGGTGACCATTGCGTC |
| p19-MscCG-A100V-R |
GACGCAATGGTCACCGGAATCGC |
| p19-MscCG-A106V-F |
GCGTCAGCTGTGATTGGTCTTG |
| p19-MscCG-A106V-R |
CAAGACCAATCACAGCTGACGC |
| p19-MscCG-A120V-F |
GGTCTTGGTGTGCAGTCGATTGTTGC |
| p19-MscCG-A120V-R |
GCAACAATCGACTGCACACCAAGACC |
| 99e-pfk-F |
CAGGAAACAGACCATGGAATTCAAAGGAGGGAAATCATGGAAGACATGCGAATTGC |
| 99e-pfk-R |
CAGGTCGACTCTAGAGGATCCCTATCCAAACATTGCCTGGGCAG |
| 99e-pyk-F |
ACAGGAAACAGACCATGGAATTCAAAGGAGGGAAATCATGGGCGTGGATAGACGAACTAAG |
| 99e-pyk-R |
CAGGTCGACTCTAGAGGATCCTTAGAGCTTTGCAATCCTTGTGT |
| pfk-pyk-1 |
CTATCCAAACATTGCCTGGGCAGTAACC |
| pfk-pyk-2 |
GGATCCTCTAGAGTCGACCTG |
| pfk-pyk-3 |
TGCCCAGGCAATGTTTGGATAGTGTTGACAATTAATCATCCGG |
| pfk-pyk-4 |
CAGGTCGACTCTAGAGGATCCCTATCCAAACATTGCCTGGGCAG |
| The bold sequences are different RBS sequences and the underlined sequences are restriction sites.
|
1.1.3 主要试剂 琼脂糖凝胶回收试剂盒、小量质粒提取试剂盒以及MultiF Seamless Assembly Mix重组酶均购于江苏康为世纪生物科技股份有限公司;EcoR Ⅰ、Hind Ⅲ等限制性核酸内切酶、DNA聚合酶及Marker均购自TaKaRa公司。
1.2 重组质粒的构建 为了加强谷氨酸棒杆菌内合成谷氨酸的前提供应,本研究过表达了谷氨酸棒杆菌来源的ppc、gltA、pfk以及pyk基因,同时为了弱化ODHC的表达,过表达了不同强度核糖体结合位点(ribosome-binding site, RBS)序列的α-酮戊二酸脱氢酶复合体(ODHC) E1亚基编码基因odhA,不同强度的RBS序列由RBS Calculator (https://salislab.net/software/)预测得到。以本实验室保存的pXMJ19和pEC-XK99E质粒为载体,以谷氨酸棒杆菌基因组为模板,通过表 1的引物分别扩增基因片段。再通过同源重组方法,构建pXMJ19和pEC-XK99E相关质粒。
pK18mobsacB-ΔalaT质粒的构建:以C. glutamicum G01基因组为模板,以引物对ΔalaT-1-F/R和ΔalaT-2-F/R分别扩增基因alaT的上下游同源臂,以EcoR Ⅰ和Hind Ⅲ限制性内切酶对本实验室保存的pK18mobsacB (pK18)质粒进行双酶切线性化。将扩增产物与线性化载体用DNA连接酶连接,将连接产物转化至E. coli JM109感受态细胞,于卡那霉素抗性平板培养,筛选转化子验证正确的接入LB培养基培养,37 ℃过夜培养取菌液抽提质粒,将重组质粒送至天霖生物科技(上海)有限公司测序以确认重组质粒pK18-ΔalaT是否构建成功[13-14]。pK18-ΔodhA、pK18-Δmscsg、pK18-MscCG-A100V质粒的构建过程与此类似。
1.3 转录组分析与基因组测序 将原始菌株C. glutamicum E01与C. glutamicum G01分别进行5 L发酵罐发酵,在菌体到达对数生长期(18 h)时取样,4 ℃、8 000 r/min离心10 min收集菌体,液氮中放置10 min后置于–80 ℃冰箱中,送至苏州金唯智生物科技有限公司进行转录组分析与基因组测序。
1.4 重组菌株的构建与表达
1.4.1 C. glutamicum基因缺失突变株的构建 C. glutamicum的基因敲除方法采用pK18mobsacB质粒进行。以alaT基因敲除为例,采用电穿孔法将pK18-ΔalaT转化[15]至C. glutamicum G01感受态细胞中,30 ℃培养2–3 h后涂布Kan抗性的BHI平板,培养48 h后利用聚合酶链式反应(polymerase chain reaction, PCR)鉴定进行第一轮筛选,挑取阳性转化子于含有Kan抗性的BHI液体培养基培养12 h后,吸取100 μL菌液涂布于含有10%蔗糖的BHI无抗性平板上,进行第二轮筛选,随机挑选转化子进行PCR鉴定,筛选到alaT基因缺失菌株G01ΔalaT后提取其基因组送至天霖生物科技(上海)有限公司测序以证明基因成功敲除。
1.4.2 过表达菌株的构建与表达 将上文构建的pXMJ19-gltA等质粒采用电穿孔法转化至C. glutamicum G01感受态细胞中,筛选正确的进行菌株命名。
将C. glutamicum G01及构建好的谷氨酸棒杆菌重组菌在氯霉素抗性BHI固体平板上划线活化,挑取单菌落接入10 mL氯霉素抗性的BHI液体小瓶中,于30 ℃、180 r/min培养12–18 h后以1%接种量接种至50 mL BHI液体培养基中,30 ℃、180 r/min培养6 h后,添加1 mmol/L终浓度的异丙基-β-d-硫代半乳糖苷(isopropyl-β-d-thiogalactoside, IPTG)诱导,继续于30 ℃、180 r/min培养12–16 h后,于4 ℃离心收集菌体。用Tris缓冲液(50 mmol/L, pH 7.5)洗涤2次菌体后,用5 mL Tris缓冲液重新悬浮,将混合液用超声破碎仪破碎后进行离心20 min (12 000 r/min, 4 ℃),收集的上清液即为粗酶液,可用于SDS-PAGE分析以及后续酶活的测定。
1.4.3 重组谷氨酸高产菌株的构建 经以上策略组合,首先对C. glutamicum G01-ΔalaT经RBS替换得到C. glutamicum G01-ΔalaT-RBS4odhA,经GDH整合得到C. glutamicum G01-ΔalaT-RBS4odhA: : An-gdh,对其进行MscCG突变得到C. glutamicum G01-ΔalaT-RBS4odhA: : An-gdh-MscCGA100V,命名为C. glutamicum AO 01,之后实现pfk、pyk以及ppc的共表达。最终得到C. glutamicum AO 01/99E-pfk-pyk/pXMJ19-ppc,将其命名为C. glutamicum AO 02。
1.5 相关酶酶活测定 丙氨酸氨基转移酶(alanine aminotransferase, ALAT)酶活力的测定:使用AT活性检测方法测定,一个单位的丙氨酸氨基转移酶活力定义为每分钟转化1 mmol/L丙氨酸所需的酶量[16]。
柠檬酸合酶酶活力的测定:通过5, 5ʹ-二硫代双(2-硝基苯甲酸)测定在412 nm下的吸光值,其与乙酰辅酶A反应会使反应体系在412 nm处的吸光值改变。一个单位的柠檬酸合酶活力定义为每分钟转化1 mmol/L乙酰-CoA所需的酶量[8]。
磷酸烯醇式丙酮酸羧化酶酶活力的测定:通过苹果酸脱氢酶偶联法测定,一个单位的磷酸烯醇式丙酮酸羧化酶酶活力定义为在30 ℃下每分钟氧化1 µmol NADH所需的酶量[17]。
α-酮戊二酸脱氢酶酶活力的测定:通过以NAD+为辅酶,测定在340 nm处ODHC催化α-酮戊二酸生成琥珀酰辅酶A反应的吸光度变化。一个单位的α-酮戊二酸脱氢酶酶活力定义为每分钟生成1 µmol NADH或NAD+ (或盐酸苯腙衍生物)所需的酶量[17]。
磷酸果糖激酶酶活力的测定:参照Blangy的方法测定340 nm处吸光值的变化,一个单位酶活力定义为每分钟转化1 µmol NAD+所需酶的量[18]。
谷氨酸脱氢酶酶活力测定:10 mmol/L辅酶NADH,0.5 mol/L硫酸铵溶液,0.1 mol/L磷酸缓冲液(pH 7.5)。按照上述反应体系配制反应溶液。酶活单位(U)定义:在上述条件下,每分钟生成1 μmol L-草铵膦所需的酶量。
丙酮酸激酶酶活力测定:参照Jetten等[19]的方法测定340 nm处吸光值的变化,一个单位酶活力定义为每分钟转化1 µmol NADH所需酶的量。
1.6 重组菌株发酵 菌株经平板划线活化后,挑取单菌落接入10 mL的BHI液体培养基,过夜培养后以1%接种量转接至10 mL种子培养基,30 ℃、180 r/min培养24 h后作为一级种子液。将一级种子全部接入至装有200 mL的种子培养基的1 L大瓶中,以相同条件培养12 h后作为二级种子液,将其全部转接至装有1.8 L发酵培养基的5 L发酵罐中。发酵条件为温度:前期30 ℃发酵,10 h (OD600约为30时)开始升温至32 ℃,后每间隔2 h升2 ℃,升至37 ℃后恒温发酵;转速600 r/min,通气量为4 vvm,pH 7.0 (流加50%氨水控制),当葡萄糖浓度降为20 g/L左右时流加80%葡萄糖溶液。需要进行诱导的菌株在发酵12 h后,OD600约为20时加入终浓度为1 mmol/L的IPTG进行诱导。
1.7 发酵过程相关参数测定 对发酵液定期取样后适当稀释,测定其在600 nm处的吸光度OD600,葡萄糖和谷氨酸的浓度采用生物传感分析仪SBA-40E测定。
2 结果与分析
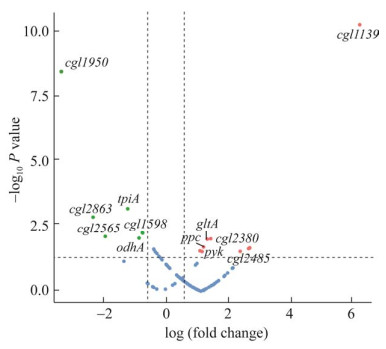
2.1 转录组及基因组数据分析 对C. glutamicum E01与C. glutamicum G01的谷氨酸合成关键途径中相关基因进行转录组差异分析。图 2中对糖酵解途径中的22个相关基因、磷酸戊糖途径中的18个相关基因以及三羧酸循环的20个相关基因进行转录水平差异分析,结果发现odhA、tpiA基因转录水平明显下调,ppc、gltA、pfk、pyk基因转录水平上调。
为了验证转录水平差异变化显著的基因是否在基因碱基处发生变化,进行了基因组全测序分析,结合实验室前期测得的重测序结果,发现基因水平上的差异,结果见表 2。对关键基因突变后的研究发现基因突变对酶活的影响不大,后续实验会在C. glutamicum G01中研究突变基因对谷氨酸合成的影响。
表 2 基因组测序差异
Table 2 Genome sequencing differences
| Genes |
Descriptions |
| gltA |
A4T, S79C, S156T, A243E, A252D |
| pfk |
V263A, S317R, E325D |
| pyk |
S327A |
| odhA |
E1220K, E1180A, N1093D, I967V, L449F, N424S, N413S, N413D, A238T, T84A |
| ppc |
Q14R, Y85H, G241D, E267G, H274R, K303E, A310E, K383T, A408S, A433T, A441E, Q542R, D549G, R634K, A704T, L773F, T829R, R840H |
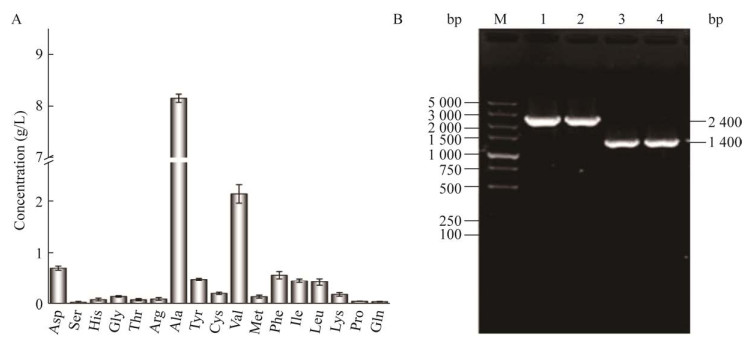
2.2 丙氨酸氨基转移酶敲除降低副产物丙氨酸含量 前期通过对C. glutamicum G01菌株发酵液的各种氨基酸含量进行测定时发现,丙氨酸为其发酵过程中的主要副产物,浓度为(8.13±0.550) g/L,其次是缬氨酸(图 3A),尝试通过对主要副产物丙氨酸合成途径中丙氨酸氨基转移酶合成基因alaT进行敲除以有效降低副产物的含量。按照1.4.1的方法进行基因敲除,PCR鉴定结果如图 3B所示,条带与目标条带1 400 bp大小一致,证明基因alaT成功敲除。
对G01-ΔalaT与G01菌株分别培养提取粗酶液后测得:G01中ALAT的比酶活为(80.0±4.43) U/mL,而在菌株G01-ΔalaT中未检测到ALAT的酶活性,进一步证明基因alaT被成功敲除。
将活化后的菌株接10 mL BHI小瓶液体培养基中,经两级种子培养后进行5 L发酵罐分批补料发酵。发现alaT敲除后与敲除前相比,丙氨酸含量由(8.13±0.550) g/L下降至(2.23±0.092) g/L,下降了72.57%,减少了副产物丙氨酸的生成,后续以G01-ΔalaT菌株为底盘进行后续的改造。
2.3 L-谷氨酸代谢流的强化
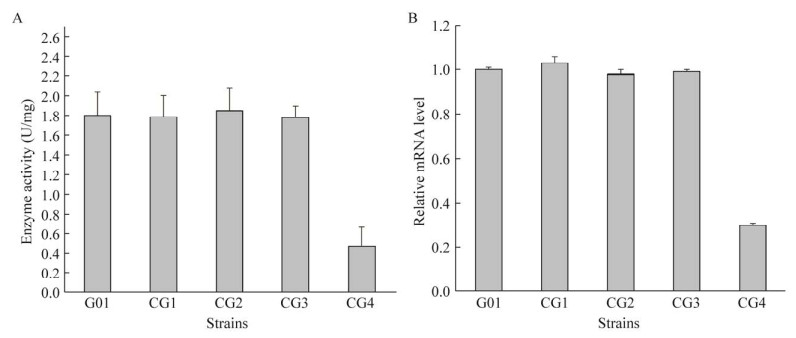
2.3.1 弱化α-酮戊二酸脱氢酶复合体活性促进谷氨酸合成 降低ODHC复合体活性是提高谷氨酸产量的一种有效方法[20],有研究称通过改变RBS序列[21]或者通过odhA抑制蛋白OdhI来降低ODHC的活性[7, 22-23]。按照1.3.2的方法构建菌株得到重组菌株CG1、CG2、CG3和CG4,在RBS预测网站上预测得到的结果显示G01原始菌的odhA基因前的RBS序列的强度为47 au,预测得到的4条RBS序列强度采取梯度降低的方式,RBS1/2/3/4的强度分别为30、20、10和5 au,同时测定这4株重组菌株与C. glutamicum G01的ODHC酶活,结果如图 4所示,重组菌CG1、CG2、CG3与原始菌相比,ODHC酶活均未有明显改变,可能是因为网站只是基于预测,并未真实地反映RBS序列对基因的调控强度,而CG4菌株的酶活有明显降低,比酶活为(0.469±0.131) U/mg,与原始菌的(1.795±0.32) U/mg相比比酶活降低了73.87%,通过对odhA转录水平的进一步测定,发现除CG4转录水平明显下降外,其余菌株转录水平未有明显变化甚至略有升高,后续研究将会使用RBS4序列进行替换研究。
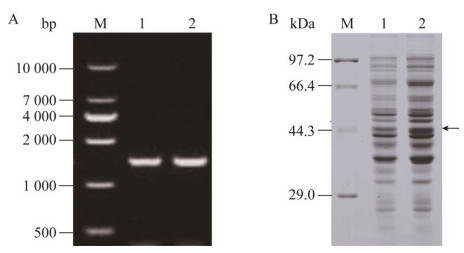
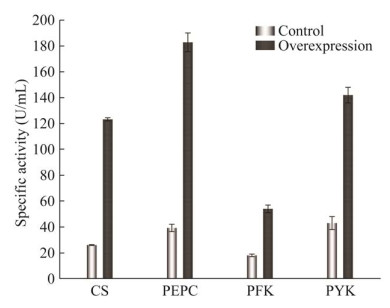
2.3.2 增加L-谷氨酸合成的前体供应菌株构建与表达 按照1.3.2中的方法进行gltA基因的过表达,PCR鉴定结果如图 5A所示,条带大小为1 314 bp,与gltA基因的大小相符,证明G01/pXMJ19-gltA构建成功。将其与对照菌株进行蛋白表达鉴定,G01/pXMJ19-gltA的CS表达量明显高于G01对照菌株,其条带大小约为42.6 kDa,说明CS成功在G01中过表达(图 5B)。对G01/pXMJ19-gltA的CS酶活测定显示为(123.26±1.24) U/mL,较对照菌G01中CS的酶活(25.97±0.37) U/mL提高了约3.8倍,即在G01/pXMJ19-gltA中gltA的过表达提高了CS的酶活水平。
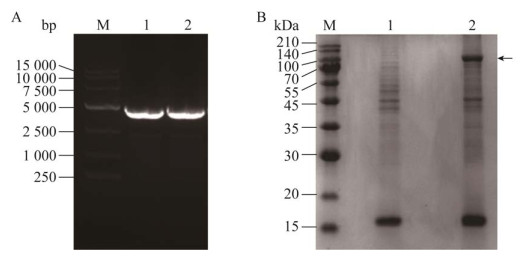
按照1.3.2中的方法进行ppc基因的过表达,PCR鉴定结果如图 6A所示,条带大小为2 760 bp,与ppc基因的大小相符,证明G01/pXMJ19-ppc构建成功。对其与对照菌株进行蛋白表达鉴定(图 6B),G01/pXMJ19-ppc的PEPC表达量明显高于G01对照菌株,其条带大小约为103.2 kDa,说明PEPC成功在G01中过表达。对G01/pXMJ19-ppc的PEPC进行酶活测定,结果显示G01/pXMJ19-ppc的PEPC酶活为(182.89±7.23) U/mL,较原始菌的(39.23±2.87) U/mL提高了约3.7倍,即在G01/pXMJ19-ppc中ppc的过表达提高了PEPC的酶活。
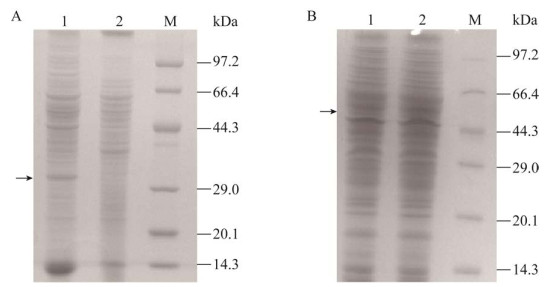
按照前述方法进行pfk、pyk基因的过表达,对重组菌株与对照菌株进行蛋白表达鉴定,重组菌株的PFK、PYK表达量明显高于G01对照菌株(图 7)。对重组进行酶活测定,重组菌G01/pEC-XK99E-pfk破碎液上清中PFK的比酶活为(54.14±0.03) U/mL,同对照的(18.23±0.01) U/mL相比提高了约2倍;重组菌G01/pEC-XK99E-pyk破碎液上清中PYK的比酶活为(142.42±0.06) U/mL,与对照的(43.68±0.05) U/mL相比提高了约2.3倍(图 8)。证明在G01中成功实现了基因的过表达并提高了PFK和PYK的酶活。
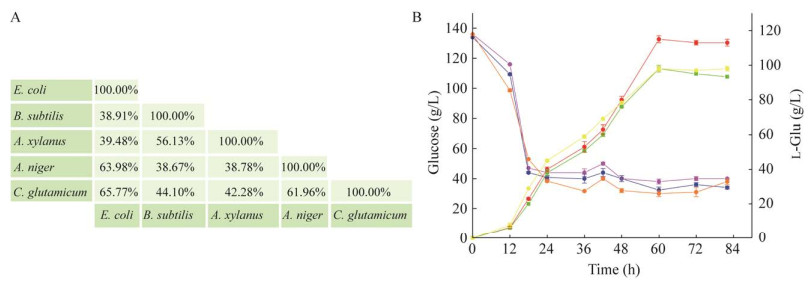
2.4 筛选不同来源谷氨酸脱氢酶提高谷氨酸合成能力 谷氨酸脱氢酶(glutamate dehydrogenase, GDH)是谷氨酸代谢的关键酶,且在有机酸测定过程中发现存在未转化完全的α-KG,因此,猜测GDH酶活水平的提高,可能会改善谷氨酸合成。本实验选取了大肠杆菌、枯草芽胞杆菌(Bacillus subtilis)、谷氨酸棒杆菌、木糖双歧杆菌(Amphibacillus xylanus)以及黑曲霉菌(Aspergillus niger)等不同来源的GDH,通过多序列比对分析得出了各自序列相似度(图 9A)。通过进行酶学性质研究,期望筛选出酶学性质稳定且酶活较高的GDH。
对不同来源的GDH进行最适pH、最适温度以及比酶活研究发现A. niger来源的GDH比酶活较高(表 3)。随后对其进行GDH过表达的菌株G01/pXMJ19-An-gdh与内源GDH过表达菌株G01/pXMJ19-Cg-gdh进行了5 L发酵罐发酵。发酵结果显示,A. niger来源GDH过表达的菌株的L-Glu产量为113.8 g/L,与对照的96.5 g/L相比提升了17.9%,而进行内源GDH过表达并未增加谷氨酸产量(图 9B),三者的耗糖速率相当。可能是因为虽然重组菌株的GDH最适温度高于原始菌,但二者最适pH相差不大且在30 ℃条件下发酵,其GDH酶活性仍高于原始菌。后续将采用A. niger来源的GDH进行基因组的整合。
表 3 不同来源GDH的酶学性质研究
Table 3 Enzymatic properties of GDH from different sources
| Source |
Optimum pH |
Optimum temperature (℃) |
Specific activity (U/mg) |
| E. coli |
7.5 |
55 |
60.0±2.8 |
| B. subtilis |
7.5 |
65 |
65.0±5.0 |
| C. glutamicum |
7.0 |
45 |
170.0±3.2 |
| A. xylanus |
8.5 |
50 |
172.0±3.6 |
| A. niger |
7.5 |
50 |
275.0±4.3 |
2.5 谷氨酸转运蛋白结构优化促进谷氨酸的外排 谷氨酸转运蛋白(MscCG)是一种机械敏感通道蛋白,有研究表明MscCG与谷氨酸的排泄有关,且仅增加MscCG的表达量并不能提高谷氨酸的产量。Nakayama等[24]对MscCG的T3跨膜区进行定点突变(A100V、A106V)和插入氨基酸的方法改变了MscCG的结构,从而提高了谷氨酸产量。由于现有研究无该转运蛋白的晶体结构,本研究选择Alphafold2.0对其三级结构进行预测,随后采用在线服务器(https://saves.mbi.ucla.edu/),通过评价参数REEAT打分的数值对转运蛋白的建模模型质量进行评价。REEAT打分为92.44 (> 85),证明建模结果可靠(图 10A)。MscCG整体结构与大肠杆菌MSCs类通道相似,但MscCG存在4个跨膜域,而MSCs仅存在3个,这使得MscCG的C端和N端细胞外环可以使通道关闭状态更加稳定[25]。随后通过Discovery Studio软件对转运蛋白及氨基酸底物进行分子对接,结果显示谷氨酸在转运出细胞时会通过转运蛋白的空腔,靠近第3个β螺旋(87A–124L)。预测谷氨酸与该螺旋上氨基酸残基产生相互作用而改变转运效率,预测得出的位点为A106V与A120V。根据上述策略,构建了菌株G01/pXMJ19-MscCGA100V、G01/pXMJ19-MscCGA106V和G01/pXMJ19-MscCGA120V。对重组菌株及G01/pXMJ19-MscCG进行5 L发酵罐发酵,发酵结果如图 10所示,增加MscCG的表达量并不能提高谷氨酸的产量。而A100V突变体较原始菌产量提高了23.58% (96.53 g/L→119.3 g/L);A106V突变体谷氨酸产量为106.7 g/L,提升了10.54%;A120V突变体较原始菌相比产量未有明显改变。可能是通道的门控阈值降低,使得渗透压引起的膜张力更容易激活通道的打开[24]。后续将采用A100V突变位点进行基因组上的整合操作。
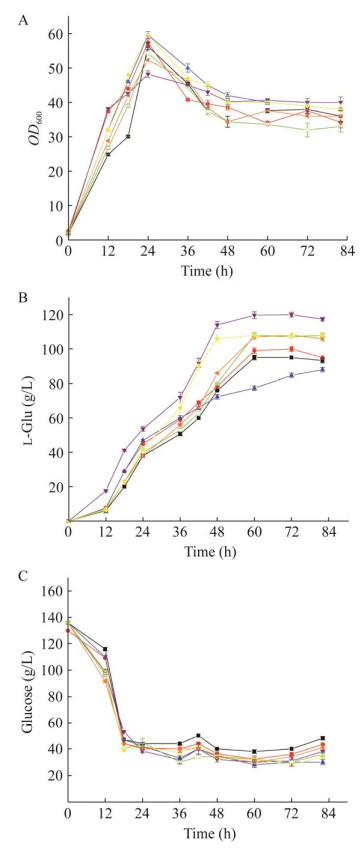
2.6 发酵验证重组菌株产谷氨酸水平 由图 11可知,发酵84 h后,菌株C. glutamicum G01谷氨酸产量达到(96.53±2.32) g/L,糖酸转化率为44.2%;C. glutamicum G01/pXMJ19-ppc与C. glutamicum G01/pXMJ19-gltA的菌株生长速率与初期的葡萄糖消耗速率与原始菌C. glutamicum G01相比有所提高,可能是PEPC酶活的提高增加了草酰乙酸的供应,强化了TCA循环,从而提高了葡萄糖消耗速率与生长速率;而CS酶活的提高直接强化了进入TCA循环起始部分的碳流量,使得菌体的生长速率与耗糖速率增加。菌株C. glutamicum G01/pXMJ19-ppc在发酵60 h处L-谷氨酸产量达到最大值(108.33±2.08) g/L,较出发菌株相比产量提高了12.2%,而菌株C. glutamicum G01/pXMJ19-gltA仅是生长速度略微变快,产量并无明显变化,L-谷氨酸产量为(90.33±2.26) g/L,说明仅通过提高CS酶活并不能提高L-谷氨酸产量[12, 26-27]。C. glutamicum G01/pEC-XK99E-pfk与C. glutamicum G01/pEC-XK99E-pyk的生长速率较原始菌相比在初期略低于原始菌,可能是pfk的过表达加强了菌体的负荷,使得初期菌体生长速率较慢,同时由于增强了EMP途径,使得葡萄糖消耗速率提高;而pyk的加强加重了菌体的负荷,且PYK控制着丙酮酸的外流量,代谢过程需要更多的糖参与[10]。C. glutamicum G01/pEC-XK99E-pfk L-谷氨酸产量为(107.66±1.89) g/L,产量提高了11.6%;C. glutamicum G01/pEC-XK99E-pyk的L-谷氨酸产量为(106.33±2.45) g/L,产量提高了10.2%。C. glutamicum G01-ΔalaT菌株在72 h处L-谷氨酸达到最大积累量(101.33±1.67) g/L,提高了5.0%。其谷氨酸产量未有明显增长,可能是因为代谢通量的改变不大。C. glutamicum G01/pXMJ19-RBS4odhA菌体生长并未受到明显影响,在发酵期间谷氨酸合成速率略高于原始菌株,于72 h处达到最大谷氨酸浓度(119.67±1.98) g/L,与原始菌株相比,L-谷氨酸积累量提高了24.0%。证明α-酮戊二酸向琥珀酰辅酶A的途径的代谢分布强弱直接影响了L-谷氨酸的合成,ODHC活性的降低能够有效地增加了L-谷氨酸的产量。
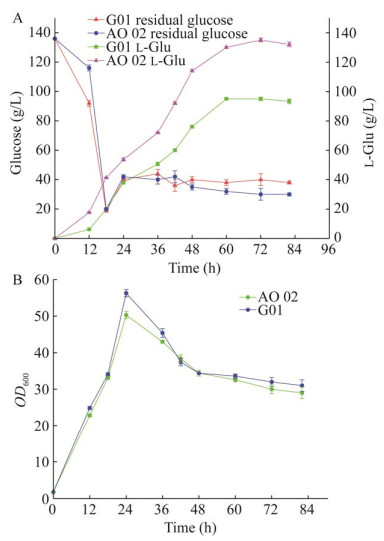
2.7 重组谷氨酸高产菌株的发酵验证 对C. glutamicum AO 02菌株进行5 L发酵罐发酵,在发酵过程中菌株的生长速率相对于原始菌略有降低,可能是菌体负荷过大导致前期耗糖速率变慢。C. glutamicum AO 02菌株发酵84 h后,其谷氨酸最大积累量为(136.33±4.68) g/L,较原始菌提高了41.2%;此外,该菌株的糖酸转化率为55.8%,较原始菌提高了11.6%,可能是因为重组菌株并没有消耗过多的糖来让自身生长而是用来合成谷氨酸。本研究构建的重组菌株,谷氨酸产量由(96.53±2.32) g/L提升至(136.33±4.68) g/L,且糖酸转化率提高了11.6% (图 12),为谷氨酸棒杆菌生产谷氨酸的代谢改造提供了理论指导,并对谷氨酸工业化菌株构建提供了新的思路。
3 讨论 L-谷氨酸广泛应用于食品、医学、农学等行业,有着巨大的应用前景。Li等[28]通过构建谷氨酸棒杆菌细胞外层成分霉菌酸合成途径中基因缺失菌株,谷氨酸产量提高了9倍。Ogata等[29]通过研究铜离子对谷氨酸棒杆菌的胁迫作用诱导产生谷氨酸,产量提高了1.3倍。陈宁等[30]通过温度控制实现细胞膜某些结构的改变,在温度转换后实现了从谷氨酸非积累型细胞向谷氨酸积累型细胞的转换,谷氨酸最高产量为181 g/L,为目前公开报道的最高水平。本研究通过系统代谢工程构建高产L-谷氨酸C. glutamicum重组菌株,对实验室前期经逐级诱变得到的谷氨酸高产菌株C. glutamicum G01进行改造,通过对底盘细胞进行副产物途径基因的敲除减少了副产物的生成,由于TCA循环起点以及α-酮戊二酸途径对谷氨酸的合成有重要意义,因此本文研究了增强磷酸烯醇式丙酮酸羧化酶、柠檬酸合酶以及α-酮戊二酸脱氢酶E1亚基弱化和谷氨酸脱氢酶酶活力对合成谷氨酸的影响,同时为了研究糖代谢速率以及丙酮酸节点对三羧酸循环中谷氨酸的影响,研究了EMP途径中的磷酸果糖激酶与丙酮酸激酶的增强对合成谷氨酸的影响,最后研究了谷氨酸外排蛋白对谷氨酸排泄的促进影响。基于以上研究,重组菌株的谷氨酸产量提高了41.2%,糖酸转化率提高了11.6%。
本文通过代谢工程手段研究了谷氨酸合成相关途径中的基因并有效地提高了其产量。但本研究还不够全面,尚未对谷氨酸分泌有重要影响的转录调控因子、辅因子供应以及发酵过程优化等方面进行相关研究。下一步会继续研究其他功能基因对谷氨酸产生的影响,进一步提高谷氨酸产量。
 2023, Vol. 39
2023, Vol. 39