
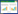


抗体成药性筛选流程及评价策略
姚江宁
,
律璎桐
,
张迎珺
,
张正平
,
徐同杰
生物工程学报  2024, Vol. 40 Issue (2): 507-516 2024, Vol. 40 Issue (2): 507-516 |

引用本文

|

近年来,各类抗体药物大量获批上市,因其具有高目标特异性、低免疫原性和长效药代动力学等优势,抗体药物在人类各个疾病治疗领域得到广泛应用[1]。随着现代生物技术的发展,抗体发现平台日渐成熟,具有特定功能的生物大分子不断被发现。但是抗体药物在研发过程中仍然有着较高的失败率[2],使得最终获批上市的药物数量十分有限,究其原因在于分子本身的一些固有特性如容易聚集、稳定性差、溶解度差等,使工艺开发、制剂开发、运输和储存等阶段面临困难,导致最终难以成药[3]。这就要求在早期分子筛选的过程中通过相关体外方法对分子的均一性、稳定性、溶解度及特异性进行评估[2],筛选优势分子,掌握候选分子的潜在风险因素,减少在后续的研发过程中因分子自身不佳的属性导致失败,该过程即为抗体药物的成药性评价。
成药性评价主要通过一系列高通量的技术手段,对抗体的一级结构、翻译后修饰、聚体和片段、电荷异质性、疏水性、构象稳定性、胶体稳定性、溶解度、黏度、蛋白质-蛋白质相互作用、非特异性相互作用、表位竞争和亲和力等方面进行多维度的分析,筛选出最佳的候选分子,确保在后续的开发中具有较为稳定的性质,从源头上保证产品的质量。本文对抗体成药性筛选和评价的不同阶段进行了系统性的分类和定义,并对筛选流程进行了优化,在各阶段设置合适的评估策略,辅助候选药物的快速开发;同时阐述了抗体发现阶段应重点评估的质量属性以及配套的高精度和高通量自动化检测技术,以期为加速抗体药物发现阶段的成药性筛选与评价过程提供思路与支持。
1 成药性筛选与评估流程传统上,抗体药物发现阶段的筛选过程被认为是漏斗型筛选[3-4],通过杂交瘤技术、展示技术(噬菌体展示、细胞表面展示、无细胞分子展示技术)、人源抗体转基因小鼠和人单B细胞测序等技术,获得数量庞大的候选序列,以与目标种属蛋白特异性结合、功能活性等评价方法作为主要评价策略进行多轮筛选。但目前越来越多的研究者将成药性评价提前至抗体药物发现阶段,有利于尽早评估和解决候选药物的可开发性问题,最终获得一个均一、稳定、安全、有效和可放大生产的候选序列,提高药物开发的成功率和效率。本文将抗体药物早期发现阶段的成药性筛选流程划分为成药性初期评价、成药性筛选和成药性评估3个阶段。成药性初期评价和筛选阶段,样品种类多、数量大,综合时间进度和有限的资源,采用倒漏斗型筛选策略,选择少量且关键的质量属性进行评价,同时配合高通量技术加快评估流程;对中后期筛选出的分子,应进行多维度且较为全面的质量属性的表征;对最终筛选出的分子,应增加加压实验等研究,以获得更为全面的分子属性。值得注意的是,在抗体药物发现阶段,成药性筛选评估没有绝对的标准对候选分子进行剔除,而是对候选分子综合评估后,打分排序(如风险等级按高、中、低排序),帮助及时了解候选药物的分子属性和潜在风险因素,掌握控制要点,便于后续工艺和制剂的开发[1, 5]。
1.1 成药性初期评价非人源的抗体候选序列(如杂交瘤筛选技术)获得后,需先构建为嵌合抗体,即对候选序列进行恒定区人源IgG1或IgG4代替,使用哺乳动物细胞系(人源胚胎肾细胞、中国仓鼠卵巢细胞等)表达;对于筛选获得的人源序列,可以直接进行质粒构建和表达;对于抗原结合片段库(fragment of antigen binding, Fab)、单链抗体可变区基因片段库(single chain variable fragment, scFv)和单域抗体库(single domain antibody, dAb)筛选得到的序列,可以制备成IgG-like的形式,也可以选择合适的表达体系进行单独表达。此阶段的表达方式一般为瞬转表达,有利于快速获得小体系样本,经高通量(正压、负压或者磁力装置)设备纯化,得到具有一定纯度的样品。
此阶段是以活性筛选策略为主,包括结合活性[酶联免疫吸附技术(enzyme linked immunosorbent assay, ELISA)、流式细胞技术(flow cytometry, FCM)]和功能活性(天然细胞、工程细胞),还可以借助高通量分子相互作用仪,如基于表面等离子共振技术(surface plasmon resonance, SPR)的BiacoreTM 8K系列仪器或者基于生物膜干涉技术(bio-layer interferometry, BLI)的Octet® BLI系列仪器,检测与目标种属靶抗原的特异性、亲和力、结合动力学和表位竞争等。在保证项目进度的前提下,可选择关键物理化学属性进行成药性初期评价,包括表达量和纯度。纯度可通过高通量技术进行检测,如体积排阻色谱-高效液相色谱(size exclusion-high performance liquid chromatography, SEC-HPLC)法、十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis, SDS-PAGE)法或毛细管凝胶电泳-十二烷基硫酸钠(capillary electrophoresis-sodium dodecyl sulfate, CE-SDS) (如Labchip®系列仪器)法等。瞬转表达量与稳转表达量具有一定的相关性,筛选高表达量的分子,同时选择不易形成聚集或降解的分子,不仅可以降低后期生产成本,还代表着具有更优的构象稳定性和胶体稳定性,也为体外活性检测奠定基础,避免因聚集体或降解物过多导致结果失真。
筛选出的序列若用于制备双特异性抗体,应在此阶段进行组装考察,评价活性和稳定性,可以通过构建质粒表达,也可以借助体外重组技术完成。评估项目包括表达量、纯度和活性,值得注意的是:纯度检测项应根据分子及其变异体性质的差异选择合适的评价策略,如可根据电荷差异通过离子色谱-高效液相色谱(ion exchange-high performance liquid chromatography, IEX-HPLC)和成像毛细管等电聚焦(imaged capillary isoelectric focusing, iCIEF)等方法进行表征,也可根据分子量大小通过SEC-HPLC和毛细管凝胶电泳法表征;筛选出的序列若用于制备抗体偶联药物(antibody-drug conjugates, ADCs),可在此阶段进行偶联,评价分子偶联效率和稳定性,如通过SEC-HPLC等方法检测聚体和片段的含量、通过疏水相互作用-高效液相色谱(hydrophobic interaction-high performance liquid chromatography, HIC-HPLC)法或质谱法检测ADC的偶联量及偶联位点的分布、通过质谱法检测小分子脱落量和通过ELISA法或质谱法检测血浆稳定性等。
1.2 成药性筛选经以上步骤筛选出的分子,借助高通量计算机模拟和分析技术,对氨基酸序列、分子结构、蛋白质和蛋白质相互作用进行初步分析和预测。针对抗体中的非人源性成分,进行人源化改造,以降低在人体内出现免疫原性的风险,保留抗体互补决定区非人源氨基酸残基,其余均替换为人抗体的相应部分,或改造暴露在抗体表面的骨架区的非人源氨基酸残基[6];对于全人源抗体库筛选出的人源抗体,需进行亲和力成熟等改造。不同的突变改造可能会影响分子稳定性,应综合考虑平衡亲和力、免疫原性和分子稳定性[7];对于其他潜在的一些问题(如等电点、脱酰胺、异构化、暴露的色氨酸和蛋氨酸、未配对半胱氨酸、糖基化或糖化等)也应给予关注和改造,如等电点过高可能会影响半衰期,等电点过低在常规的工艺过程中易产生聚集;互补决定区的脱酰胺、异构化导致分子降解或者活性降低;氧化可能会影响活性,也可能引发聚集体的产生;可变区的糖基化或糖化位点,可能会影响活性和稳定性等等[3]。
改造后的分子通过轻重链的组合配对,产生一系列的变体,数量一般在100个左右,通过哺乳动物细胞瞬转表达和高通量纯化(依据活性实验需求进行内毒素控制),评价表达量、纯度和生物学活性,去除极差分子后,开展成药性筛选评价。成药性筛选阶段的目标为对各分子的固有属性进行评估和排序,如电荷变异体含量、疏水性、热稳定性、不同粒径聚集体、自相互作用、非特异性相互作用、与不同种属抗原的亲和力、表位差异、抗体可结晶段(fragment crystallizable, Fc)功能等[1]。最终挑选出具有较低异质性、较高稳定性、较长半衰期、高产量和可工艺放大的抗体,筛选出的分子数量一般小于5个。本阶段成药性筛选所用到的检测技术,应具备样品用量少、通量高、分析快速等特点。若候选分子具有特殊属性,可在此阶段增加初步的稳定性评估,以确保样品在检测、存储、动物给药过程中的稳定,如室温稳定性、2−8 ℃稳定性、冻融稳定性等。若候选分子计划用于制备高浓度剂型,可以增加溶解度预测实验,随着蛋白浓度的升高,相较于低浓度溶液,蛋白与蛋白之间的作用力增强,但传统的溶解度测定方法对蛋白消耗量大,故可选用在较低浓度中预测溶解度的方法[8-9]。
1.3 成药性评估对于最终筛选出的几个候选分子,进行进一步的可开发性评估。表达阶段扩大瞬转体系,纯化阶段增加精纯步骤,并严格控制内毒素含量,为早期的动物体内药代动力学(pharmacokinetics, PK)、药效动力学(pharmacodynamics, PD)以及毒理研究储备样品。“成药性筛选阶段”进行的成药性表征方法应再次检测和确认,另外还应进行加压实验的考察,有助于快速地暴露分子的风险位点及变化趋势,考察对抗体特异性和功能活性的影响程度,考察分子的耐受性和可及性,评估敏感条件,预测长期稳定性,如设置40 ℃高温稳定性,纳米抗体可进行100 ℃高温稳定性考察。通过翻译后修饰、聚集体和降解物含量、酸碱变异体含量、ADC中小分子脱落量的变化,发现分子潜在的翻译后修饰位点、降解位点及变化趋势;还可以设置强酸、强碱、氧化和光照等加压实验;对于高浓度制剂的开发,在前期溶解度预测实验中,由于高的稀释因子,难以准确推导高浓度制剂真实的溶解度和黏度,故在此阶段,可以开展目标浓度的浓缩和过浓缩,使用常用缓冲体系,添加尽可能少的制剂辅料,观察药物在浓缩过程中的特性,包括但不限于蛋白含量、聚集体和降解物的含量、粒径的尺寸分布和分散度、黏度、热稳定性、储存稳定性以及在高浓度下与血清的相融性等。最佳配方条件的探索,则应在后期制剂开发中进行[1]。
2 抗体的分子属性及相关检测方法 2.1 一级结构的确认与翻译后修饰抗体的一级结构是指从N端到C端的氨基酸残基的线性序列。一级结构的确认通常包括分子量、氨基酸序列覆盖率、N/C端氨基酸序列分析等。其中,抗体分子量检测的目的是确认是否有片段错配或缺失,包括完整分子量、还原分子量、脱糖分子量和脱糖还原分子量,均采用基于质谱的检测方法,其中脱糖相关分子量可用特异性的N/O糖酶将N/O糖切除后检测。对于双抗或多抗,目标分子与错配分子的分子量差异很小,精确分子量对于检测错配比例尤为重要。抗体的氨基酸序列覆盖率是将样品变性还原和烷基化后,加入特异性酶进行酶切,经液相色谱-质谱检测后与理论序列对比得到的。常用的酶包括胰蛋白酶、胞内蛋白酶、糜蛋白酶等。
抗体的翻译后修饰(post-translational modification, PTM)是指蛋白质经翻译后发生的化学加工,其中糖基化是抗体最常见的翻译后修饰,主要有N-糖基化和O-糖基化。其中,N-糖基化为最普遍、最常见的糖基化类型。抗体的N-糖基化发生在抗体Fc的CH2区上的天冬酰胺-X-丝氨酸/苏氨酸位点,X为除脯氨酸外的任何氨基酸。糖基化可增加抗体的稳定性,维持蛋白结构完整。另外,糖基化还能提高抗体的溶解度。在生物学活性方面,糖基化修饰会影响抗体Fc结合各种抗体可结晶段γ受体(fragment crystallizable gamma receptor, FcγR)的亲和力,例如,去岩藻糖化可显著提高抗体与抗体可结晶段γ受体Ⅲa (fragment crystallizable gamma receptor Ⅲa, FcγRⅢa)的亲和力,进而增强抗体依赖细胞介导的细胞毒作用(antibody-dependent cell-mediated cytotoxicity, ADCC)[10]。糖基化的表征一般分为3个水平进行,分别为完整抗体、糖基化肽段和游离寡糖水平。完整抗体水平可利用质谱的方法观察到主要糖型的分布。抗体片段水平的分析是将抗体进行酶解,产生的小肽经色谱或电泳分离后进行质谱分析,可得到氨基酸序列、糖基化位点等信息。游离寡糖的分析则是通过糖苷酶的酶切将N-糖苷游离出来,再通过液相色谱-质谱法进行寡糖的定性和定量分析。
除糖基化外,常见的翻译后修饰还有甲硫氨酸的氧化、天冬酰胺的脱酰胺、N末端环化和C末端赖氨酸丢失等。这些修饰可能会影响抗体的构象和电荷,表现出不同比例的酸碱峰。翻译后修饰与氨基酸序列覆盖率检测方法相似,将样品变性还原和烷基化后,加入特异性酶进行酶切,经液相色谱-质谱检测后,根据序列比对结果,计算翻译后修饰的位点和比例。二硫键也是抗体中常见的1种翻译后修饰,它是2个半胱氨酸残基的巯基基团发生氧化反应形成的共价键[11-12]。但二硫键并不稳定,可被还原为游离的巯基,而游离巯基可能会降低抗体的稳定性。常用的游离巯基定量方法为埃尔曼试剂检测法,质谱法也可用来表征游离巯基的位点。
2.2 聚体和片段抗体的聚体含量和片段化程度都属于产品的关键质量属性(critical quality attributes, CQAs),在整个药物的研发和生产过程中需密切监控。聚体是抗体分子间通过共价键或非共价等分子间作用力形成的聚合物[13],片段化则是抗体中的共价键被破坏的过程,可能由酶催化或化学降解等方式造成;双特异性抗体在生产过程中会产生包括同源二聚体、缺少1条轻链的抗体、半抗体、重链聚合物、轻链聚合物、游离重链和轻链等副产物;ADC的裸抗通过不同键合方式与连接子-有效载荷(linker-payload)相连接,其中巯基偶联是目前主流的偶联方式之一,裸抗链间二硫键的破坏以及具有一定疏水性的有效载荷(payload)可能导致片段和聚体的产生。聚体的形成可能引起人体免疫原性反应,对患者的安全造成威胁,而片段会降低药物的生物学活性[14-15]。
目前,体积排阻-高效液相色谱法是测定抗体聚体和片段组分最常用的方法,该方法在天然条件下检测抗体的组分,条件较为温和,可真实反映抗体在溶液中的状态。十二烷基硫酸钠-聚丙烯酰胺凝胶电泳或毛细管凝胶电泳则在变性条件下检测聚体和片段,其中,毛细管凝胶电泳法可被开发为较高通量的检测方法,且分离度较高,可直接定量。例如Labchip®系列仪器采用了微流控技术,将样品与嵌入染料混合后进行电泳分析,且兼容96孔板与384孔板,极大地提高了检测速度。以上2种电泳方法可鉴别体积排阻-高效液相色谱法未观察到的碎片,常与体积排阻-高效液相色谱法共同用于产品的研发和生产过程中。除此之外,反相-高效液相色谱法(reversed phase-high performance liquid chromatography, RP-HPLC)可根据抗体片段的疏水性差异进行分离,与质谱联用可以直接鉴定裂解位点,但与毛细管凝胶电泳法相比,分辨率较低,可作为研究阶段的辅助方法。
2.3 电荷异质性抗体在生产和存储过程中会发生脱酰胺化、氧化、异构化和片段化等多种翻译后修饰或化学降解,双特异性抗体的错配副产物若存在电荷差异,这些会影响抗体电荷基团的数量或结构,造成抗体电荷分布的异质性和等电点(isoelectric point, pI)的改变[16]。其中,等电点值较低的电荷变体为酸性电荷变体,等电点值较高的为碱性电荷变体。酸碱电荷变体的产生会对抗体的生物学活性、药代动力学、免疫原性等造成一定的影响,因此在整个生产工艺中,都应对抗体的电荷异质性进行监测。离子交换色谱法已被广泛用于电荷变体的检测,该方法对仪器要求不高,液相色谱就可满足检测需求,且可放大到制备级用于分离制备酸碱性电荷变体进行后续研究[17]。毛细管等电聚焦(capillary isoelectric focusing, cIEF)、成像毛细管等电聚焦技术以高灵敏度、自动化的优势,也成为分析电荷变异体的主要方法。毛细管区带电泳(capillary zone electrophoresis, CZE)可根据电荷和流体动力学半径分离抗体的电荷变体,其优势在于高通量,且可以与质谱仪在线耦合。
2.4 疏水性抗体的疏水性与抗体的聚集和沉淀倾向有关。疏水相互作用-高效液相色谱是基于疏水性差异进行分离的技术,在高盐状态下分子内部部分疏水氨基酸暴露,抗体表面的疏水区域与固定载体上的疏水配体相互作用,使抗体分子吸附至固定相,然后降低流动相的盐浓度以实现洗脱。由于ADC分子偶联了非极性的小分子,疏水性增大,且不同药物抗体偶联比(drug-to-antibody ratio, DAR)的分子疏水性也有差异,可通过疏水相互作用-高效液相色谱分离以检测ADC分子的DAR值及DAR值分布。除了疏水相互作用-高效液相色谱外,反相-高效液相色谱也可基于抗体的疏水性差异进行分离,但与疏水相互作用-高效液相色谱的温和条件相比,反相-高效液相色谱法的测试条件常为低酸碱度(potential of hydrogen, pH)、高温和高有机相条件,抗体可能因为处于变性条件中,无法表征和收集到维持天然空间构象的蛋白馏分,因此疏水相互作用-高效液相色谱是目前最常用的检测疏水性的方法。ADC中小分子残留可以通过反相-高效液相色谱或串联质谱的方法进行检测。
2.5 构象稳定性抗体在极端温度或pH值、化学变性剂等条件下可能通过解折叠而失去高级结构,进而影响抗体的功能。抗体构象稳定性的检测方法包括差示扫描量热法(differential scanning calorimetry, DSC)、差示扫描荧光法(differential scanning fluorimetry, DSF)、等温化学变性和圆二色谱法(circular dichroism, CD)等。差示扫描量热法和差示扫描荧光法都是通过对蛋白溶液进行梯度升温,检测热变性温度[18]。差示扫描量热法是测量蛋白变性过程中吸收的热量,而差示扫描荧光法是基于抗体解折叠引起疏水和芳香残基的暴露,进而导致外在或内在荧光的增加[19]。差示扫描荧光法的通量更高,但可能会受到分子内芳香残基数目的影响[20]。等温化学变性是在化学变性条件下,测量折叠蛋白和变性蛋白的能量差(吉布斯自由能),该方法不基于温度的变化,但要求蛋白处于可逆变性条件下。圆二色谱是基于蛋白质分子对左、右圆偏振光的吸收差异,分别通过远紫外区(180−250 nm)和近紫外区(250−320 nm)反映蛋白的二级、三级结构[21]。
2.6 胶体稳定性完整折叠的蛋白在溶液中会有自身相互作用,主要由折叠后分子表面的疏水基团和电荷分布决定。若自身相互作用力表现为排斥力,溶液则会表现出较好的胶体稳定性;若表现为吸引力,就会产生聚集倾向。聚体检测最常用的方法是SEC-HPLC,可根据分子量分离物质,其中高分子量物质被最先洗脱,为获得更多信息,可在SEC-HPLC中加入光散射检测器,如多角度光散射(multi-angle laser light scattering, MALS)、直角光散射(right-angle light scattering, RALS)和低角度光散射(low-angle light scattering, LALS)检测器,用于测定组分的分子量(molecular weight, MW),但该方法不适用于过大的聚集体。动态光散射法(dynamic light scattering, DLS)使用斯托克斯-爱因斯坦方程,通过颗粒运动引起的散射光强度波动推断颗粒尺寸,可应用于0.3−10 000 nm内的粒子检测,对蛋白溶液进行梯度升温,通过监测颗粒尺寸,得到抗体的聚集温度(the aggregation temperature, Tagg)[22-23]。
2.7 溶解度在制剂开发过程中,需采用适当的方法预测抗体的溶解度。此外,由于皮下注射在临床应用中的便利性,许多抗体希望开发为皮下注射制剂。但抗体药物剂量大,而通常皮下注射的体积不超过2 mL,使得许多抗体制剂需要制成较高的蛋白浓度。抗体的溶解度较低通常是由于暴露的疏水基团或电荷分布的相互作用引起的。聚乙二醇(polyethylene glycol, PEG)浓度梯度沉淀法可推测抗体的表观溶解度,且可开发为高通量方法[8]。但该方法仅能预测抗体的溶解度,也应考虑直接评估抗体在高浓度下的溶解度以进一步确认。
除此之外,渗透第二维里系数(the osmotic second virial coefficient, B22)和扩散相互作用系数(the diffusion interaction parameter, kD)与抗体的高浓度行为有关,B22和kD的正值和负值分别代表排斥力或吸引力,且kD已被证明与B22相关,能够在低浓度条件下表征分子间作用力的大小。B22和kD可分别基于静态光散射(static light scattering, SLS)和动态光散射原理(DLS)检测。DLS相关的高通量设备,如DynaProTM Plate Reader可兼容96孔板、384孔板、1 536孔板,样品量低至4 µL。
2.8 黏度制剂的黏度是产品工艺开发中的另一个关键因素,黏度的具体数值可通过黏度计测出。对于拟开发为高浓度制剂的抗体,可使用微量黏度测量技术,在节省样品的前提下,准确测定不同浓度下的溶液黏度,如m-VROC® Ⅱ半自动超微量黏度计/流变仪仅需要15 μL样品即可检测黏度。此外,通过DLS方法测量的扩散相互作用系数(kD)也显示出与黏度相关,可作为黏度预测的指标之一。
2.9 蛋白质-蛋白质相互作用/非特异性相互作用除了抗体本身分子属性外,蛋白质-蛋白质相互作用也会影响抗体的溶解度、黏度和聚集。另外,部分抗体的快速清除可能与其在体内的非特异性结合相关。因此在开发阶段,应对蛋白质-蛋白质相互作用进行评估。交叉相互作用色谱法(cross-interaction chromatography, CIC)可表征抗体与偶联在色谱柱上抗体的相互作用,洗脱时间的延长与非特异性吸附相关[24];ELISA的方法可预测抗体与杆状病毒颗粒(baculovirus particles, BVP)的非特异性作用,并在一定程度上预测抗体的快速清除[25];基于亲和捕获的自相互作用纳米粒子光谱(affinity-capture self-interaction nanoparticle spectroscopy, AC-SINS)则是一种高通量的检测方法,将抗人的捕获抗体包被在金纳米颗粒上,固定抗体后,通过等离子体波长的红移来检测抗体的自相互作用。Estep等[26]分别用AC-SINS和HIC-HPLC法对一组抗体进行了表征,并证明了AC-SINS和HIC-HPLC法的良好相关性。
2.10 亲和力亲和力用于评价抗体的功能,即与靶点分子的结合能力,也是成药性评价中重要的一个环节。对于有明确靶点的抗体,在嵌合抗体、人源化以及进一步的结构改造等阶段,均需要进行亲和力检测,并根据结合和解离速率,判断抗体与靶点作用的动力学特征。而抗体除了与靶点分子结合外,其Fc端与FcγR等受体、新生儿Fc受体(the neonatal Fc receptor, FcRn)、补体(complement 1q, C1q)的亲和力也与其功能密切相关,其中,FcγR等受体的结合强度可表征抗体的ADCC、抗体依赖细胞介导的细胞吞噬作用(antibody-dependent cell-mediated phagocytosis, ADCP);FcRn与C1q分别反映了抗体的半衰期、补体依赖的细胞毒作用(complement dependent cytotoxicity, CDC)。
亲和力可通过酶联免疫吸附法、表面等离子共振法、生物膜光干涉技术等方法进行评估。酶联免疫吸附法是将抗原或抗体固定在聚苯乙烯微孔板内,利用酶标记的抗体或抗原与之孵育,显色剂显色后,根据终点颜色深浅评估结合强弱。表面等离子共振,是在全内反射条件下,入射光使薄金层等离子体发生共振,导致反射光在某一特定角度(即SPR角)能量低至几乎为零,分子在传感器上的结合、解离会导致金膜附近的折光率改变,仪器将这一变化转变为电信号输出,可实时检测抗原抗体的结合、解离情况。Kitazawa等[27]利用表面等离子共振技术辅助揭示了双抗艾美赛珠(Emicizumab)结合凝血因子IX/IXa和凝血因子X/Xa的详细机制。生物膜光干涉技术是基于生物分子结合到传感器表面形成的生物膜,传感器的光波使生物膜发生干涉现象,并以相位移动的形式被反射,从而反映抗原抗体的结合、解离。目前,基于表面等离子共振的BiacoreTM 8K系列仪器以及基于生物膜光干涉技术的Octet® BLI系列仪器通过增加进样针的个数、样品仓内可容纳孔板的数量、适配的孔板类型等方式,逐渐可以满足更高通量的检测、筛选需求。
以上3种方法均是目前检测亲和力的通用方法,其中表面等离子共振和生物膜光干涉技术是实时、动态、无标记的检测,相较于终点法ELISA而言,检测更加灵敏、准确,可以更好地反映抗体的功能。
3 总结与展望成药性评价作为一种在药物发现阶段被广泛认可的候选分子筛选方式,已经成为抗体药物研发过程中的关键一环。不同成药性筛选阶段,选择符合筛选目的的评估策略,不仅能理解分子目标属性,还能大幅减少筛选时间,降低冗余数据堆砌,最终加快筛选流程和提高研发成功率。本文系统性地将成药性筛选评估策略进行了分类和定义,在不同阶段设置相对应的筛选指标,结合一系列高精度和高通量的自动化技术手段,分阶段、多维度评估分子属性,从大量候选分子中筛选出最优的目标序列。经此过程筛选获得的分子不仅具有更优秀的自身质量属性,还能够通过最终的可开发性评估建立分子的成药性指纹图谱,为后续的工艺开发、制剂开发等过程提供指导与支持。
经过几十年的发展,抗体药物在各治疗领域获得巨大进展,但仍存在一些未满足的临床治疗需求,需要进一步对药物靶点、疾病机制进行深入研究,设计开发新型药物分子形式,如近几年的双(多)特异性抗体、抗体偶联药物等。新型药物分子设计的多样性和复杂性更加需要系统性、科学性的成药性评价策略,如何认识和解析新型药物分子属性,如何选择合适的筛选评价方法,如何设定打分排序标准,都是巨大的挑战。借助新一代的信息技术,可扩充新型药物成药性评价手段,如通过人工智能技术建立深度学习模型可对新型药物结构和性能进行预测和评估排序,确保成药性评估能够成为“分子知识”和“分子理解”之间坚实的桥梁,扩大候选分子的设计空间,提高产品开发效率,加快抗体药物高质量发展。
| [1] |
JARASCH A, KOLL H, REGULA JT, BADER M, PAPADIMITRIOU A, KETTENBERGER H. Developability assessment during the selection of novel therapeutic antibodies[J]. Journal of Pharmaceutical Sciences, 2015, 104(6): 1885-1898. DOI:10.1002/jps.24430
|
| [2] |
ZURDO J. Developability assessment as an early de-risking tool for biopharmaceutical development[J]. Pharmaceutical Bioprocessing, 2013, 1(1): 29-50. DOI:10.4155/pbp.13.3
|
| [3] |
BAILLY M, MIECZKOWSKI C, JUAN V, METWALLY E, TOMAZELA D, BAKER J, UCHIDA M, KOFMAN E, RAOUFI F, MOTLAGH S, YU Y, PARK J, RAGHAVA S, WELSH J, RAUSCHER M, RAGHUNATHAN G, HSIEH M, CHEN YL, NGUYEN HT, NGUYEN N, et al. Predicting antibody developability profiles through early stage discovery screening[J]. mAbs, 2020, 12(1): 1743053. DOI:10.1080/19420862.2020.1743053
|
| [4] |
ZHANG WJ, WANG H, FENG N, LI YF, GU JJ, WANG ZZ. Developability assessment at early-stage discovery to enable development of antibody-derived therapeutics[J]. Antibody Therapeutics, 2023, 6(1): 13-29. DOI:10.1093/abt/tbac029
|
| [5] |
JAIN T, SUN TW, DURAND S, HALL A, HOUSTON NR, NETT JH, SHARKEY B, BOBROWICZ B, CAFFRY I, YU Y, CAO Y, LYNAUGH H, BROWN M, BARUAH H, GRAY LT, KRAULAND EM, XU YD, VÁSQUEZ M. Biophysical properties of the clinical-stage antibody landscape[J]. Proceedings of the National Academy of Sciences of the United States of America, 2017, 114(5): 944-949.
|
| [6] |
SAFDARI Y, FARAJNIA S, ASGHARZADEH M, KHALILI M. Antibody humanization methods-a review and update[J]. Biotechnology and Genetic Engineering Reviews, 2013, 29(2): 175-186. DOI:10.1080/02648725.2013.801235
|
| [7] |
DOEVENDANS E, SCHELLEKENS H. Immunogenicity of innovative and biosimilar monoclonal antibodies[J]. Antibodies (Basel, Switzerland), 2019, 8(1): 21.
|
| [8] |
CHAI Q, SHIH J, WELDON C, PHAN S, JONES BE. Development of a high-throughput solubility screening assay for use in antibody discovery[J]. mAbs, 2019, 11(4): 747-756. DOI:10.1080/19420862.2019.1589851
|
| [9] |
WALKER JM. Molecular Biology[M]. vol 2313. New York: Humana New York, 2022: 57-113.
|
| [10] |
MAO C, NEAR R, ZHONG XM, GAO WD. Cross-species higher sensitivities of FcγRIIIA/FcγRIV to afucosylated IgG for enhanced ADCC[J]. Antibody Therapeutics, 2021, 4(3): 159-170. DOI:10.1093/abt/tbab016
|
| [11] |
LIU HC, MAY K. Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function[J]. mAbs, 2012, 4(1): 17-23. DOI:10.4161/mabs.4.1.18347
|
| [12] |
李明, 吴佩泽. 基于质谱技术的二硫键定位分析方法研究进展[J]. 质谱学报, 2021, 42(6): 985-994. LI M, WU PZ. Research progress on disulfide bond assignment analysis based on mass spectrometry[J]. Journal of Chinese Mass Spectrometry Society, 2021, 42(6): 985-994 (in Chinese). |
| [13] |
PANG KT, YANG YS, ZHANG W, HO YS, SORMANNI P, MICHAELS TCT, WALSH I, CHIA SA. Understanding and controlling the molecular mechanisms of protein aggregation in MAb therapeutics[J]. Biotechnology Advances, 2023, 67: 108192. DOI:10.1016/j.biotechadv.2023.108192
|
| [14] |
LUNDAHL MLE, FOGLI S, COLAVITA PE, SCANLAN EM. Aggregation of protein therapeutics enhances their immunogenicity: causes and mitigation strategies[J]. RSC Chemical Biology, 2021, 2(4): 1004-1020. DOI:10.1039/D1CB00067E
|
| [15] |
VLASAK J, IONESCU R. Fragmentation of monoclonal antibodies[J]. mAbs, 2011, 3(3): 253-263. DOI:10.4161/mabs.3.3.15608
|
| [16] |
WANG W, SINGH S, ZENG DL, KING K, NEMA S. Antibody structure, instability, and formulation[J]. Journal of Pharmaceutical Sciences, 2007, 96(1): 1-26. DOI:10.1002/jps.20727
|
| [17] |
DU Y, WALSH A, EHRICK R, XU W, MAY K, LIU HC. Chromatographic analysis of the acidic and basic species of recombinant monoclonal antibodies[J]. mAbs, 2012, 4(5): 578-585. DOI:10.4161/mabs.21328
|
| [18] |
NIESEN FH, BERGLUND H, VEDADI M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability[J]. Nature Protocols, 2007, 2(9): 2212-2221. DOI:10.1038/nprot.2007.321
|
| [19] |
SVILENOV H, MARKOJA U, WINTER G. Isothermal chemical denaturation as a complementary tool to overcome limitations of thermal differential scanning fluorimetry in predicting physical stability of protein formulations[J]. European Journal of Pharmaceutics and Biopharmaceutics: Official Journal of Arbeitsgemeinschaft Fur Pharmazeutische Verfahrenstechnik e V, 2018, 125: 106-113.
|
| [20] |
le BASLE Y, CHENNELL P, TOKHADZE N, ASTIER A, SAUTOU V. Physicochemical stability of monoclonal antibodies: a review[J]. Journal of Pharmaceutical Sciences, 2020, 109(1): 169-190. DOI:10.1016/j.xphs.2019.08.009
|
| [21] |
HAWE A, KASPER JC, FRIESS W, JISKOOT W. Structural properties of monoclonal antibody aggregates induced by freeze-thawing and thermal stress[J]. European Journal of Pharmaceutical Sciences, 2009, 38(2): 79-87. DOI:10.1016/j.ejps.2009.06.001
|
| [22] |
MAHLER HC, FRIESS W, GRAUSCHOPF U, KIESE S. Protein aggregation: pathways, induction factors and analysis[J]. Journal of Pharmaceutical Sciences, 2009, 98(9): 2909-2934. DOI:10.1002/jps.21566
|
| [23] |
PAUL M, VIEILLARD V, JACCOULET E, ASTIER A. Long-term stability of diluted solutions of the monoclonal antibody rituximab[J]. International Journal of Pharmaceutics, 2012, 436(1/2): 282-290.
|
| [24] |
JACOBS SA, WU SJ, FENG YQ, BETHEA D, O'NEIL KT. Cross-interaction chromatography: a rapid method to identify highly soluble monoclonal antibody candidates[J]. Pharmaceutical Research, 2010, 27(1): 65-71. DOI:10.1007/s11095-009-0007-z
|
| [25] |
HÖTZEL I, THEIL FP, BERNSTEIN LJ, PRABHU S, DENG R, QUINTANA L, LUTMAN J, SIBIA R, CHAN P, BUMBACA D, FIELDER P, CARTER PJ, KELLEY RF. A strategy for risk mitigation of antibodies with fast clearance[J]. mAbs, 2012, 4(6): 753-760. DOI:10.4161/mabs.22189
|
| [26] |
ESTEP P, CAFFRY I, YU Y, SUN TW, CAO Y, LYNAUGH H, JAIN T, VÁSQUEZ M, TESSIER PM, XU YD. An alternative assay to hydrophobic interaction chromatography for high-throughput characterization of monoclonal antibodies[J]. mAbs, 2015, 7(3): 553-561. DOI:10.1080/19420862.2015.1016694
|
| [27] |
KITAZAWA T, ESAKI K, TACHIBANA T, ISHII S, SOEDA T, MUTO A, KAWABE Y, IGAWA T, TSUNODA H, NOGAMI K, SHIMA M, HATTORI K. FactorVIIIa-mimetic cofactor activity of a bispecific antibody to factors IX/IXa and X/Xa, emicizumab, depends on its ability to bridge the antigens[J]. Thrombosis and Haemostasis, 2017, 117(7): 1348-1357. DOI:10.1160/TH17-01-0030
|