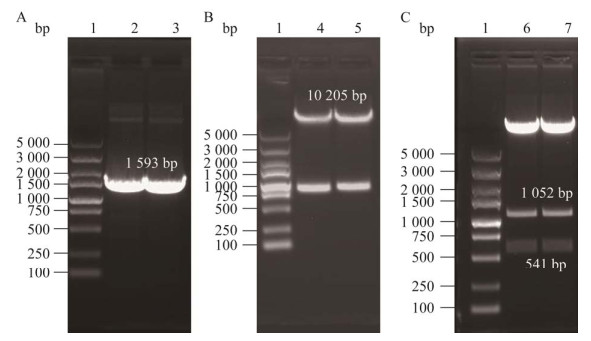
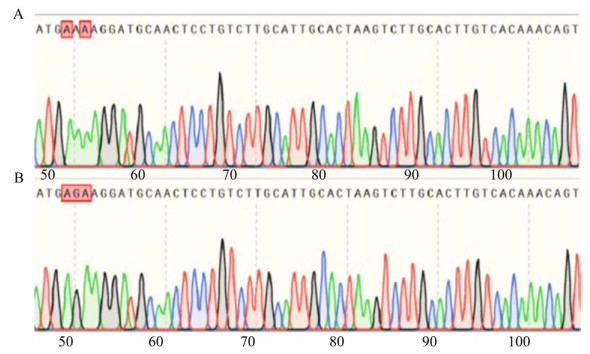
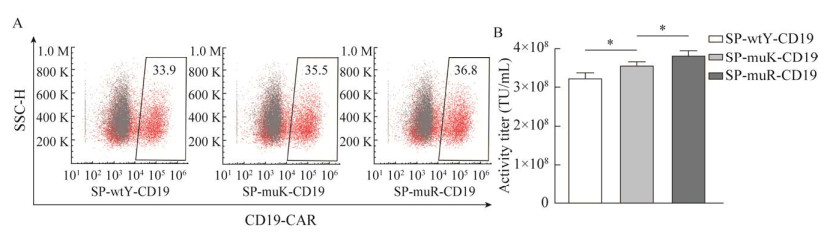
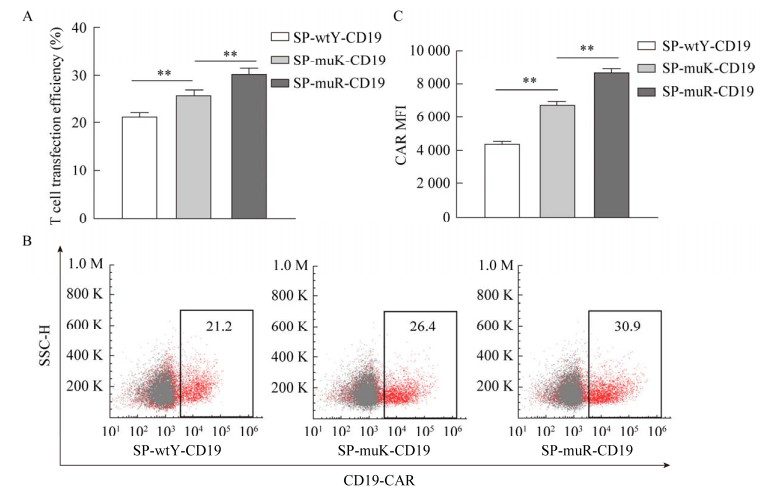
| [1] |
JOHNSON LA, JUNE CH. Driving gene-engineered T cell immunotherapy of cancer[J]. Cell Research, 2017, 27(1): 38-58. DOI:10.1038/cr.2016.154
|
|
| [2] |
JUNE CH, SADELAIN M. Chimeric antigen receptor therapy[J]. New England Journal of Medicine, 2018, 379(1): 64-73. DOI:10.1056/NEJMra1706169
|
|
| [3] |
BRUDNO JN, KOCHENDERFER JN. Recent advances in CAR T-cell toxicity: mechanisms, manifestations and management[J]. Blood Reviews, 2019, 34: 45-55. DOI:10.1016/j.blre.2018.11.002
|
|
| [4] |
DEPIL S, DUCHATEAU P, GRUPP SA, MUFTI G, POIROT L. 'Off-the-shelf' allogeneic CAR T cells: development and challenges[J]. Nature Reviews Drug Discovery, 2020, 19(3): 185-199. DOI:10.1038/s41573-019-0051-2
|
|
| [5] |
GUMBER D, WANG LD. Improving CAR-T immunotherapy: overcoming the challenges of T cell exhaustion[J]. eBioMedicine, 2022, 77: 103941. DOI:10.1016/j.ebiom.2022.103941
|
|
| [6] |
ARMENTEROS JJA, TSIRIGOS KD, SØNDERBY CK, PETERSEN TN, WINTHER O, BRUNAK S, von HEIJNE G, NIELSEN H. ignalP 5.0 improves signal peptide predictions using deep neural networks[J]. Nature Biotechnology, 2019, 37(4): 420-423. DOI:10.1038/s41587-019-0036-z
|
|
| [7] |
HUANG Q, PALMER T. Signal peptide hydrophobicity modulates interaction with the twin-arginine translocase[J]. mBio, 2017, 8(4): e00909-00917.
|
|
| [8] |
DANIEL W, LEE, MD. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial[J]. The Lancet, 2015, 385(9967): 517-528. DOI:10.1016/S0140-6736(14)61403-3
|
|
| [9] |
MAUDE SL, LAETSCH TW, BUECHNER J, RIVES S, BOYER M, BITTENCOURT H, BADER P, VERNERIS MR, STEFANSKI HE, MYERS GD, QAYED M, de MOERLOOSE B, HIRAMATSU H, SCHLIS K, DAVIS KL, MARTIN PL, NEMECEK ER, YANIK GA, PETERS C, BARUCHEL A, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia[J]. New England Journal of Medicine, 2018, 378(5): 439-448. DOI:10.1056/NEJMoa1709866
|
|
| [10] |
PARK JH, RIVIÈRE I, GONEN M, WANG XY, SÉNÉCHAL B, CURRAN KJ, SAUTER C, WANG YZ, SANTOMASSO B, MEAD E, ROSHAL M, MASLAK P, DAVILA M, BRENTJENS RJ, SADELAIN M. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia[J]. The New England Journal of Medicine, 2018, 378(5): 449-459. DOI:10.1056/NEJMoa1709919
|
|
| [11] |
TURTLE CJ, HAY KA, HANAFI LA, LI D, CHERIAN S, CHEN XY, WOOD B, LOZANSKI A, BYRD JC, HEIMFELD S, RIDDELL SR, MALONEY DG. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-specific chimeric antigen receptor-modified T cells after failure of ibrutinib[J]. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 2017, 35(26): 3010-3020. DOI:10.1200/JCO.2017.72.8519
|
|
| [12] |
PORTER DL, LEVINE BL, KALOS M, BAGG A, JUNE CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia[J]. The New England Journal of Medicine, 2011, 365(8): 725-733. DOI:10.1056/NEJMoa1103849
|
|
| [13] |
KOCHENDERFER JN, DUDLEY ME, FELDMAN SA, WILSON WH, SPANER DE, MARIC I, STETLER-STEVENSON M, PHAN GQ, HUGHES MS, SHERRY RM, YANG JC, KAMMULA US, DEVILLIER L, CARPENTER R, NATHAN DA N, MORGAN RA, LAURENCOT C, ROSENBERG SA. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells[J]. Blood, 2012, 119(12): 2709-2720. DOI:10.1182/blood-2011-10-384388
|
|
| [14] |
KOCHENDERFER JN, DUDLEY ME, KASSIM SH, SOMERVILLE RPT, CARPENTER RO, STETLER-STEVENSON M, YANG JC, PHAN GQ, HUGHES MS, SHERRY RM, RAFFELD M, FELDMAN S, LU L, LI YF, NGO LT, GOY A, FELDMAN T, SPANER DE, WANG ML, CHEN CC, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor[J]. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 2015, 33(6): 540-549. DOI:10.1200/JCO.2014.56.2025
|
|
| [15] |
李帆, 张琴星, 童祥文, 田高辉, 顾力行, 徐瑶. 不同信号肽对嵌合抗原受体T细胞杀伤作用的影响研究[J]. 中国癌症杂志, 2022, 32(2): 142-151.
LI F, ZHANG QX, TONG XW, TIAN GH, GU LX, XU Y. A study on influence of different signal peptides on anti-tumor effect of chimeric antigen receptor (CAR) T cells[J]. China Oncology, 2022, 32(2): 142-151 (in Chinese).
|
|
| [16] |
GOLUBOVSKAYA V. CAR-T cells targeting immune checkpoint pathway players[J]. Frontiers in Bioscience-Landmark, 2022, 27(4): 121. DOI:10.31083/j.fbl2704121
|
|
| [17] |
ROGOSIC S, GHORASHIAN S. CAR-T cell therapy in paediatric acute lymphoblastic leukaemia-past, present and future[J]. British Journal of Haematology, 2020, 191(4): 617-626. DOI:10.1111/bjh.17153
|
|
| [18] |
LIN WY, WANG HH, CHEN YW, LIN CF, FAN HC, LEE YY. Gene modified CAR-T cellular therapy for hematologic malignancies[J]. International Journal of Molecular Sciences, 2020, 21(22): 8655. DOI:10.3390/ijms21228655
|
|
| [19] |
HONG MH, CLUBB JD, CHEN YY. Engineering CAR-T cells for next-generation cancer therapy[J]. Cancer Cell, 2020, 38(4): 473-488. DOI:10.1016/j.ccell.2020.07.005
|
|
| [20] |
STERNER RC, STERNER RM. CAR-T cell therapy: current limitations and potential strategies[J]. Blood Cancer Journal, 2021, 11(4): 69. DOI:10.1038/s41408-021-00459-7
|
|
| [21] |
JANDA CY, LI J, OUBRIDGE C, HERNÁNDEZ H, ROBINSON CV, NAGAI K. Recognition of a signal peptide by the signal recognition particle[J]. Nature, 2010, 465(7297): 507-510. DOI:10.1038/nature08870
|
|
| [22] |
SARAOGI I, SHAN SO. Molecular mechanism of co-translational protein targeting by the signal recognition particle[J]. Traffic (Copenhagen, Denmark), 2011, 12(5): 535-542. DOI:10.1111/j.1600-0854.2011.01171.x
|
|
| [23] |
SADELAIN M, BRENTJENS R, RIVIÈRE I. The basic principles of chimeric antigen receptor design[J]. Cancer Discovery, 2013, 3(4): 388-398. DOI:10.1158/2159-8290.CD-12-0548
|
|
| [24] |
MAUS MV, JUNE CH. Making better chimeric antigen receptors for adoptive T-cell therapy[J]. Clinical Cancer Research, 2016, 22(8): 1875-1884. DOI:10.1158/1078-0432.CCR-15-1433
|
|
| [25] |
WANG CM, WU ZQ, WANG Y, GUO YL, DAI HR, WANG XH, LI X, ZHANG YJ, ZHANG WY, CHEN MX, ZHANG Y, FENG KC, LIU Y, LI SX, YANG QM, HAN WD. Autologous T cells expressing CD30 chimeric antigen receptors for relapsed or refractory Hodgkin lymphoma: an open-label phase I trial[J]. Clinical Cancer Research: an Official Journal of the American Association for Cancer Research, 2017, 23(5): 1156-1166. DOI:10.1158/1078-0432.CCR-16-1365
|
|
| [26] |
RAMOS CA, BALLARD B, ZHANG HM, DAKHOVA O, GEE AP, MEI ZY, BILGI M, WU MF, LIU H, GRILLEY B, BOLLARD CM, CHANG BH, ROONEY CM, BRENNER MK, HESLOP HE, DOTTI G, SAVOLDO B. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes[J]. The Journal of Clinical Investigation, 2017, 127(9): 3462-3471. DOI:10.1172/JCI94306
|
|
| [27] |
BROWN CE, ALIZADEH D, STARR R, WENG LH, WAGNER JR, NARANJO A, OSTBERG JR, KILPATRICK J, SIMPSON J, KURIEN A, PRICEMAN SJ, WANG XL, HARSHBARGER TL, D'APUZZO M, RESSLER JA, JENSEN MC, BARISH ME, CHEN MK, PORTNOW J, FORMAN SJ, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy[J]. The New England Journal of Medicine, 2016, 375(26): 2561-2569. DOI:10.1056/NEJMoa1610497
|
|
| [28] |
AHMED N, BRAWLEY VS, HEGDE M, ROBERTSON C, GHAZI A, GERKEN C, LIU EL, DAKHOVA O, ASHOORI A, CORDER A, GRAY T, WU MF, LIU H, HICKS J, RAINUSSO N, DOTTI G, MEI ZY, GRILLEY B, GEE A, ROONEY CM, et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma[J]. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 2015, 33(15): 1688-1696. DOI:10.1200/JCO.2014.58.0225
|
|
 2024, Vol. 40
2024, Vol. 40